Mit Gnathostoma spinigerum präsentiere ich heute zum ersten Mal in der "weekly Nematode"-Serie (ich sollte die Serie vielleicht mit etwas understatement in "The annual Nematode" umbenennen) einen spirurinen Nematoden. Spiruina bildet eine Schwestergruppe der Tylenchina (die beiden letzen Nematoden der Woche) und Rhabditina (meist freilebend; z.B. C. elegans). Diese Klade (Spirurina wird häufig auch einfach "Clade 3" genannt) wude erstmals 1998 entdeckt und umfasst die beiden früher auf Ordnungs-Ebene geführten Kladen Spirurida in Ascaridida.
Besonders interessant ist die Gattung Gnathostoma tatsächlich durch ihre phylogenetische Position. Da ich zurzeizeit Hilfe von einem Diplomanden habe, der sich um die Phylogeny der Gattung Anguillicola und um eine bessere Auflösung des Baums für die basalen Spirurina kümmern soll, gibt es hier eine Neuheit- einen Gastpost "meines" Diplomanden Dominik Laetsch:
Grund für unser Interesse an der Gattung Gnathostoma ist also -wie gesagt- zum einen die geringe phylogenetische Distanz der SSU-rDNA-Sequenz dieses Nematoden zu jener unseres "Lieblings-Wurms" Anguillicola crassus und die Tatsache, dass die Familien Gnathostomatidae und Aguillicolidae aufgrund jener Daten den basalen Zweig der Spirurina darzustellen scheinen [1], [2]. Bestimmte Vertreter der Gattung Gnathostoma werden uns daher auch als "Out-group" in der phylogenetischen Analyse des Genus Anguillicola dienen (was das Thema meiner Diplomarbeit darstellt).
Der Genus Gnathostoma gliedert sich in ca. 9 mehr oder weniger gut von einander abgegrenzten Arten, welche wie alle Spirurina parasitisch leben. Davon sind mindestens drei als humanpathogene Arten beschrieben: G. turgidum, G. doloresi und G. spinigerum. Der Lebenszyklus von G. spinigerum ist bekannt und umfasst die Entwicklung der L1-Larve zum, für den Endwirt infektiösen, dritten Larvenstadium in zwei aquatischen Zwischenwirten (Copepoden des Genus Cyclops bzw. Fische/ Amphibia/ Mollusca) und die anschliessende Entwicklung zum Adultus im Endwirt (Mammalia; oft Feliden, Caniden und Suiden). Paratenische Wirte stellen vor allem Fische und Vögel dar. Im Endwirt durchbohrt G. spinigerum nach der oralen Aufnahme infizierten Gewebes die Magenwand und bereist als Larva migrans für die nächsten 3 Monate dessen Gewebe und innere Organ bis er zurückkehrt und sich an die mucosale Magenwand heftet. Dort entwickelt er sich für weitere 6 Monate bis er beginnt unembryonierter Eier zu produzieren, welche durch mit dem Kot ausgeschieden werden. Im Wasser embryonieren diese Eier, und mit der Azfnahme durch den ersten Zwischenwirt ist der Kreislauf geschlossen.
Das klinische Bild wird unter dem Begriff Gnathostomiasis [3] zusammengefasst und kann sowohl bei Feliden, Caniden als auch Hominiden letal verlaufen. Erstmals beschrieben wurde dieses Krankheitsbild 1835 anhand eines Kadavers eines jungen Tigers des Londoner Zoos. In Menschen entwickelt sich G. spinigerum nicht zum Adultus sondern migriert durch das Körpergewebe für bis zu 10- 12 Jahre, wobei zwischen kutaner und viszeraler Larva migrans unterschieden wird. Ersteres beschreibt nur Hautexzesse, bedingt durch mechanische Zerstörung des Gewebes und Produktion von Proteasen, Hyaluronidasen und Hemolysin durch den Parasiten, sowie durch die Immunreaktion des Wirtes. Letzteres umfasst die selben Faktoren jedoch in Organen wie der Leber und dem ZNS. Diese Scäden führen zu einer Mortalität von 8-25 % der betrofenen Patienten. Desweiteren gibt es drei dokumentierte Fälle von intrauteriner Transmission.
Bis auf Thailand, wo sie die häufigste parasitäre Erkrankung des ZNS darstellt, ist sie jedoch selbst in ihren endemischen Gebieten (Japan, Korea, Laos, Malaysia, Taiwan, Thailand, Mexico, Ecuador) relativ selten.
FAZIT: ein gewiefter Parasit der nur mittels serologischem Test und Biopsien nachgewiesen werden kann. Vorsicht ist geboten bei "zu frischem" Fisch, dreckigem Wasser (Copepoden) und nicht garem Geflügel.
References:
[1] S. A. NADLER, R. A. CARRENO, H. MEJÍA-MADRID, J. ULLBERG, C. PAGAN, R. HOUSTON and J.-P. HUGOT (2007). Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology, 134 , pp 1421-1442
doi:10.1017/S0031182007002880
[2] Martina Wijova, Frantisek Moravec, Ales Horak, Julius Lukes, Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences, International Journal for Parasitology Volume 36, Issue 9, , August 2006, Pages 1067-1075.
doi:10.1017/S0031182007002880
[3] Moore DAJ, McCrodden J, DeKumyoy P, Chiodini PL. Gnathostomiasis: an emerging imported disease. Emerg Infect Dis [serial online] 2003 Jun
Samstag, 27. September 2008
Vergleichende Entwicklungsgenomik- Zwei Paper und massig Fehlinterpretationen
Evo-Devo (Evolutionary developmental biology) lässt sich am besten mit "Evolutionäre Entwicklungsbiologie" ins Deutsche übersetzen. Der Forschungszweig basiert darauf, dass natürliche Selektion oft nicht unmittelbar auf den Genotyp eines Individuums wirken kann, sondern auf den Phänotyp. Dieser Phänotyp wird durch ein komplexes genetisches Programm bei der Entwicklung des Organismus produziert. Bei einem solchen Entwicklungsprogramm sind die Mengen an Proteinen oder RNAs entscheidend; so können gewisse Schwellenwerte dieser Moleküle Zellschicksale beeinflussen und den Phänotyp bestimmen.
Eines der grundsätzlichsten Dogmen von Evo-Devo ist daher, dass Unterschiede in der Genexpression während der Entwicklung für unterschiedliche Phänotypen verantwortlich sind. Diese Expressionsunterschiede wiederum werden durch Unterschiede in nicht kodierenden, den Genen vorgelagerten (cis-)regulatorischen Sequenzen (hauptsächlich Promotoren und Enhancer) oder in den Protein-Sequenzen von übergeordneten Transkriptionsfaktoren verursacht (oder wiederum in der Expression dieser Transkriptionsfaktoren).
Evo-Devo will daher in die bisweilen postulierte Lücke zwischen Macro- und Microevolution vorstoßen. Gerade Experten auf dem Gebiet der Entwicklungsbiologie räumen mitunter die Möglichkeiten saltatorischer Evolution ein. Das heißt, sie halten Makromutationen für möglich, die in einem einzigen Mutationsschritt starke Änderungen des Phänotyps (sogenannte "Hopeful Monster") produzieren. Dies soll hauptsächlich durch Änderungen an wichtigen "Schaltstellen"-Transkriptinsfaktoren in den Entwicklungsprogrammen geschehen. Diese Sichtweise wird von der Mehrzahl der Evolutionsbiologen -bis auf Darwin selbst zurückgehend- abgelehnt. Die konventionelle Theorie besagt, dass Veränderungen durch graduelle Mutationen erfolgen, die sich über Generationen akkumulieren. Einzelne Wissenschaftler auf dem Gebiet der evolutionären Entwicklungsbiologie versuchen also mitunter Modifikationen an den bestehenden Fundamenten der Evolutionsbiolgie zu erreichen, was auf heftige Kritik (auch aus den eigenen Reihen) stößt. Wahrscheinlich ist Evo-Devo daher eines der lebendigsten Forschungsgebiete in der aktuellen Biologie.
Die von mir im Folgenden besprochenen Studie untersuchen beide dieses zentrale Dogma von Evo-Devo genauer und lassen meines Erachtens auch Schlüsse zur genannten Kontroverse zu.
Cretekos et al. benutzen dazu die Unterschiede im Wachstum der Vorderextremitäten zwischen Carollia perspicillata (einer Fledermaus) und der Maus. Beide Taxa stammen aus unterschiedlichen Ordnungen der Säugetiere Chiroptera (=Fledertiere) beziehungsweise Rodentia (=Nagetiere), teilen also einen gemeinsamen Vorfahren vor etwa 80-100 Millionen Jahren.
Die Studie betrachtet Veränderungen in Prx1, einem Transkriptionsfaktor, der durch klassische entwicklungsbiologische Methoden als wichtig in der Extremitätenentwicklung identifiziert wurde. Die Kollegen stellten beim Verglich der Gen-Sequenzen aus Maus und Fledermaus nur einen nicht-synonymen (in das Protein übersetzten)Unterscheid fest. Dieser Unterschied befindet sich in einem Bereich des Genes, der ohnehin wenig konserviert ist, und nicht mit der typischen Funktion des Traskriptionsfaktors in Verbindung gebracht wird.
Unterschiede in der Expression von Prx1 konnten in der späten Entwicklung der Vorderextremitäten festgestellt werden, in der Fledermaus war der Transkriptionsfaktor speziell im Bereich der Handwurzelknochen stärker exprimiert. Dieses Ergebnis korreliert gut mit dem zu diesem Zeitpunkt verstärkt auftretenden Längenwachstum in der Fledermaus.
Cretekos et al. betrachteten weiterhin also einen Enhancer "stromaufwärts" des Gens, der ebenfalls mit klassischen Methoden identifiziert worden war. Dieser Enhancer enthält zwei Bereiche die zwischen Nager und Fledertier relativ konserviert sind, in diesen wurde dann die Funktion vermutet. Um dies zu testen konnte die Gruppe die jeweilige Enhancer-Region an ein Reportegen koppeln und in Mäuse einbringen, dabei wurde stärkere Reporter-expression beim Chiroptera-Enhancer beobachtet.
Doch damit nicht genug, der Gruppe gelang es schließlich das Fledermaus-Kontrollelement in die Maus einzubringen, so dass es die Expression des Prx1-Gens steuert. Die entsprechenden Mäuse zeigten tatsächlich ein verstärktes Wachstum der Vorderextremitäten.
Die Studie konnte so eindrucksvoll das zentrale Dogma der evolutionären Entwicklungsbiologie bestätigen und weiterhin zeigen, dass die zugrunde liegenden Veränderungen im untersuchten Fall auf Enhancer-Elementen basieren.
Auch die Studie von Prabhakar et al. beschäftigt sich mit der Funktion von Enhancern, die betreffende genomische Region war aber mit anderen Mitteln identifiziert worden. Dabei wurden komplett sequenzierten Genome von Wirbeltieren nach konservierten, nichtkodierenden Sequenzen durchsucht. Aus diesen Sequenze wurden wiederum jene identifiziert, die in der Menschlichen Linie (entgegen des allgemeinen Trends) evolvieren. Diese Vorgehen basiert auf der Tatsache, dass Elemente mit einer bestimmten Funktion weniger evolvieren als funktionslose Elemente, ändern sie allerdings ihre Funktion erfolgt die Evolution sogar schneller als dies unter Neutralität (Funktionslosigkeit) der Fall wären. Die identifizierte Region liegt im Intron eines Gens, das mit der Funktion des Endosoms in Verbindung gebracht wird. Weier "stromäbwärts" befindet sich wieder ein Transkriptionsfaktor, dessen Wirkung die Entwicklung der Gliedmaßen beeinflusst. Die Expression welcher Gene genau der mögliche Enhancer beeinflusst ist also noch nicht geklärt.
Zur Untersuchung der Funktion der Enhancer-Region benutzten die Kollegen also wieder ein Reporterassay (ß-Galactosidase). Sie brachten die postulierten Kontrollelemente aus Rhesusaffen (Macaca mulatta), Schimpansen (Pan troglodytes) und dem Menschen (Homo sapiens) (Divergenz vor 6 bzw. 25 Mya) gekoppelt an das Reportergen, in Mäuse ein. So konnten die Forscher zeigen, dass die menschlichen Elemente im Vergleich zu denen aus beiden anderen Primaten, verstärkt Gene beim Wachstum der Extremitäten anschalten. Sie konnten so die durch den Genom-Verglich identifizierten Unterschiede bestätigen: Der Zustand und die Wirkung des Enhancers sind sich in nicht-menschlichen Affen ähnlich und entsprechen daher wahrscheinlich dem Zustand im gemeinsamen Vorfahren.
Mit sehr cleveren Experimenten bewiesen die Kollegen, dass genau die 13 Basen Unterschied im Menschen im Vergleich zu Schimpanse und Rhesusaffe den Unterschied ausmachen. Sie konstruierten Fragmente aus den beiden "Vierbeinern" in denen nur die betroffenen 13 Unterschiede eingebracht waren. Diese Konstrukte hatten die gleiche Wirkung wie das original-menschliche Element.
Die Studie konnte so, aufbauend auf dem zuvor beschriebenen zentralen Dogma von Evo-Devo, zeigen, dass durch Genomvergleiche die betreffenden Elemente identifiziert werden können. Ein denkbarer Name für ein solches Vorgehen ist "Vergleichende Entwiklungsgenomik" oder im Englischen "Comparative developmental genomics".
Weiter demonstrieren beide Studien eindrucksvoll die evolutionären Möglichkeiten für graduelle Veränderungen in Entwicklungsprogrammen. In beiden Beispielen konnte durch Veränderung einzelner Basen in regulativen Bereichen Unterschiede in der Genexpression erzeugt werden. Da es sich um Variationen in einzelnen Basenpaaren (SNPs) handelt, die langsam und nacheinander ins Genom einfließen, verlangen die hier beobachteten Unterschiede geradezu nach Gradualismus.
Fee hat vor einigen Wochen auf dem Blog Science-meets-spciety ebenfall über das letztere Paper geschrieben und ich möchte einige Fehler in diesem (auch auf Researchblogging erschienenen) Post abschließend korrigieren. Ich hoffe so eine Diskussion anzuregen, falls es im deutschsprachigen Raum genügend Interesse gibt.
S-M-S:
Wir teilen bis zu 98% der codierenden DNA mit unseren nächsten Verwandten, den Schimpansen (Pan troglodytes) und doch unterscheiden wir uns markant von ihnen.
Das stimmt nicht! Eine der grundlegendsten Entdeckungen der letzten Jahre waren Unterschiede in der Kopienzahl einzelner Gene im Menschlichen Genom. 2007 wurde so (Hauptsächlich durch den Vergleich der Genome von Venter und Watson) offensichtlich, dass einzelne Menschen sich in 2-3% ihres Genoms unterscheiden. Der Unterschied zu unseren nächsten interspeziefschen Verwandten dürfe daher mindestens 5% betragen.
S-M-S:
Der größte Teil der DNA in menschlichen Zellen ist nicht kodierend und wurde, als man dies entdeckte, fälschlicherweise als Junk-DNA (Abfall-DNA) verschrien.
Diese Sichtweise ist grundsätzlich falsch! Es wurden seit der Entdeckung nicht-kodierender Bereiche schon versucht Funktionen für diese für eine höhere Organisation zu postulieren. Vergleicht man aber unterschiedliche Organismen (z.B. der Zwiebel oder des Salamanders) mit der des Menschen oder der Kugelfische (Tetraodontidae; anderes Extrem) fällt schnell auf dass eine höhere Komplexität nicht mit der Genomgröße korreliert.
Die in Frage stehenden regulatorischen Elemente machen einen winzigen Teil des Genoms aus.
S-M-S:
Dabei stiessen sie auf einen Abschnitt von 546 Basenpaaren Länge, der sich seit der Entwicklung der Wirbeltiere nur wenig verändert hatte. Jedoch hatten sich in der relativ kurzen Zeit von 6 Millionen Jahren, seit sich die Entwicklungszweige von Mensch und Schimanse trennten, 16 Veränderungen etabliert, die alle in einem Abschnitt von 81 Basenpaaren clusterten. So etwas ist für einen genetischen Detektiv ein eindeutiges Indiz, dass weitere Untersuchungen gewinnbringend sein könnten.
Die Forscher wussten im Gegensatz zum Schreiber dieser Zeilen um die Existenz von Enhancen. Andernfalls hätten sie den entsprechenden Bereich nicht als solchen identifizieren können. In der besprochenen Studie wurden nicht zum ersten Mal Selektion auf einen nicht-kodierenden Bereich nachgewiesen. Ähnliche Fehlinterpretationen und die übertriebene Darstellung von Neuheiten (wissenschaftlichen Revolutionen) in der Wissenschaftsberichterstattung veranlassen auch beispielsweise Kreationisten regelmäßig zu ähnlichen Dummheiten.
S-M-S:
[...]so aktivierten alle die Expression von Genen in den Augen, Ohren und in den embryonalen Kiemenbögen, die später den Kiefer bilden.
Die Expression eines Reportergens! Dies ist im Vergleich zu den anderen Fehlern aber eher zweitrangig.
S-M-S:
Ein neuer Teilbereich der entwicklungsgenetischen Forschung, der bestimmt noch viele Überraschungen und neue Erkenntnisse birg.
Quatsch! "Vergleichende Entwicklungsgenomik" ist zwar ein recht neuer boomender Bereich der Entwicklunsbiologie, erfunden wurde er aber in diesem Paper nicht. Die Studie bestätigt vielmehr experimentell die Validität der zugrundeliegenden in silico Analysen.
Ich hab hier mal die jährliche Zähl an Veröffentlichungen, die "Comparative developmental genomics" im Volltext (Pub-med) erwähnen geplotet:
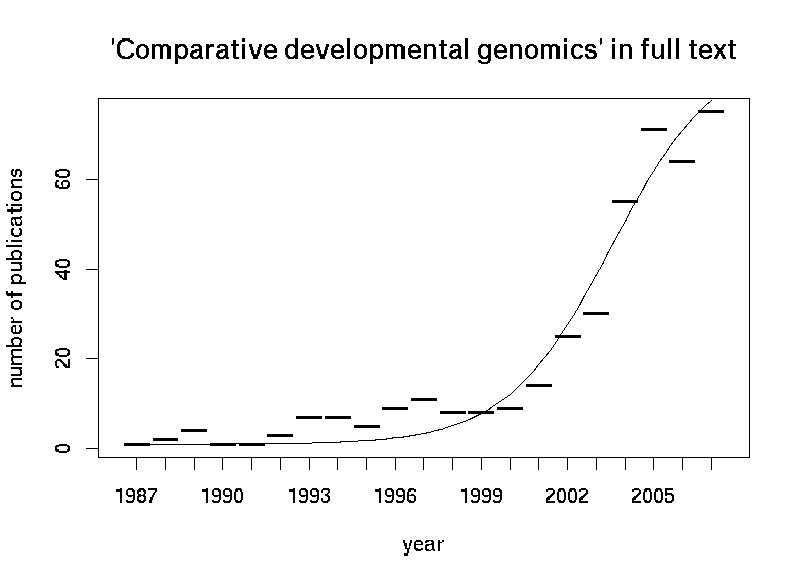
Dabei fällt auf, dass das erste Paper bereits aus dem Jahre 1988 stammt, als eigentlich noch keine Genome zum Vergleich bereitstanden. Veröffentlichungen vor dem Jahre 2001 können also wahrscheinlich als "Hintergrundrauschen" oder "Vorahnung" interpretiert werden, wirkliche "Comparative genomics" in dem heute etablierten Sinn waren damals noch kaum möglich. Ab diesem Zeitpunkt wurden dann anhand der vorhandenen Daten die betreffenden Methoden entwickelt. Prabhakar et al. haben also zwar eine schöne Studie angefertigt, mitnichten aber das "Teilgebiet" erfunden.
Kommentare/ Kritik erwünscht, ich bin kein Entwicklungsbiologe und habe sicher selbst Fehler gemacht!
________________________________________________________________________________________________________________
C. J. Cretekos, Y. Wang, E. D. Green, J. F. Martin, J. J. Rasweiler, R. R. Behringer (2008). Regulatory divergence modifies limb length between mammals Genes & Development, 22 (2), 141-151 DOI: 10.1101/gad.1620408
S. Prabhakar, A. Visel, J. A. Akiyama, M. Shoukry, K. D. Lewis, A. Holt, I. Plajzer-Frick, H. Morrison, D. R. FitzPatrick, V. Afzal, L. A. Pennacchio, E. M. Rubin, J. P. Noonan (2008). Human-Specific Gain of Function in a Developmental Enhancer Science, 321 (5894), 1346-1350 DOI: 10.1126/science.1159974
Freitag, 26. September 2008
Nochmal Aua: E.O. Wilson in der NYTimes
Schämen muss man sich eigentlich nicht wenn, man so nen Fehler macht. Fee ist in guter Gesellschaft, wie ich in diesem Post auf Myrmecos erfahren habe:
;-)
Given the antiquity of the lineage, the temptation to view Martialis as an ur-ant of sorts is strong. E.O. Wilson certainly felt that way when interviewed for a recent NYTimes article:With due respect to Wilson, such a view is a mistake. Martialis has over 120 million years’ separation since the ur-ant, plenty of time to develop along its own trajectory.Dr. Wilson…is trying to contain his excitement: the 14,001st ant species has just been discovered in the soils of a Brazilian forest. He steamrolls any incipient skepticism about the ant’s uniqueness — the new species is a living coelacanth of ants, a primitive throwback to the first ant, a wasp that shed its wings and assigned all its descendants to live in earth, not their ancestral air. The new ant is so alien, Dr. Wilson explains, so unlike any known to earthlings, that it will be named as if it came from another planet.
;-)
Dienstag, 23. September 2008
Keine Diskussion erwünscht?
Ein Post aus unerfreulichem Anlass... zunächst die Vorfälle in chronologischer Abfolge ohne Bewertung.
Durch den deutschen Arm von Research-Blogging bin ich Ende letzter Woche auf einen Post auf dem Blog Science-meets-Society aufmerksam geworden. Fee und Uli schreiben dort auf leicht verständliche Art über ein sehr breites Spektrum wissenschaftlicher Veröffentlichungen.
Bei einem kurzen Blick auf nur einen Post über den Fund einer neuen Ameisengattung (Paper) ist mir dann aber ein gewaltiger Fehler aufgefallen:
Mein erster Kommentar (genau wiedergeben kann ich diesen nicht, dazu später mehr) war aus Zeitmangel etwas barsch:
Meine Meinung dazu:
Im deutschsprachigen Raum fehlt bislang eine Diskussionskultur auf Wissenschaftsblogs. Researchblogging ist sicher ein guter Ansatz dies zu ändern. Kritik an Blogposts muss auch in ungeschminkter Form erlaubt sein (dieser Post ist übrigens genauso grottenschlecht, und wird von Argent23 "mit Samthandschuhen" kritisiert). Der Name des Blogs (Science-meets-society; science communication) ist im Zusammenhang mit der Löschung unerwünschter Kommentare geradezu lächerlich.
Ich bin ein großer Fan guter Vereinfachungen von komplexen Zusammenhängen in populärwissenschaftlichen Texten zur Evolutionsbiologie und allgemein von Wissenschaftsjournalismus. Allerdings braucht man um den betreffenden Sprachgebrauch zu beherrschen eine fundierte Kenntnis des Fachgebiets und einige Erfahrung (wegen letzterem lesen sich manche meiner Posts vielleicht auch etwas kryptisch). Gerade die Vereinfachung macht die Sache eben nicht leichter.
Zum thematischen Ursprung des Disputs noch soviel: Wer sich weiter informieren möchte kann dies auf deutschen Blogs noch nicht (falls doch bin ich dankbar für nen Link). Ich werd mich drum kümmern sobald ich etwas mehr Zeit hab! Über Fallstricke im Verständnis von Evolutionsbiologie kann man sich solange super (besser als ich das wahrscheinlich jemals hinbekommen werde) bei T. Tyan Gregory informieren.
Durch den deutschen Arm von Research-Blogging bin ich Ende letzter Woche auf einen Post auf dem Blog Science-meets-Society aufmerksam geworden. Fee und Uli schreiben dort auf leicht verständliche Art über ein sehr breites Spektrum wissenschaftlicher Veröffentlichungen.
Bei einem kurzen Blick auf nur einen Post über den Fund einer neuen Ameisengattung (Paper) ist mir dann aber ein gewaltiger Fehler aufgefallen:
Phylogenetische Analysen haben nun gezeigt, dass es sich bei Martialis wahrscheinlich um den ältesten bekannten Vorfahren der modernen Ameisen handelt, der in einem so stabilen Lebensraum wie dem Regenwaldboden, lange Zeit überdauern konnte. Dies wirft ein völlig neues Licht auf die Evolution und die Entwicklung von außergewöhnlichen Lebensweisen, heute bekannter Ameisen.
Mein erster Kommentar (genau wiedergeben kann ich diesen nicht, dazu später mehr) war aus Zeitmangel etwas barsch:
Aua! Fehler! Eine moderne (heute lebende) Ameisengattung kann niemals der Vorfahre aller heute lebender Ameisengattungen sein.Nach Lesen des Papers hab ich dann etwa folgendes geschrieben:
Die Autoren lehnen sich im "absract" weit aus dem Fenster indem sie- trotz sehr dürftiger Hinweise- spekulieren, die gefundene Ameisenart könnte ihrem mit allen anderen Ameisen gemeinsamen Vorfahren sehr ähnliche sein. In den "conclusions" relativieren sie dann allerdings ihre Hypothese wieder; " The exact nature of the ancestral ant remains uncertain given that the propensity for repeated evolution of an epigaeic lifecycle".Überraschenderweise waren diese beiden Kommentare Tags darauf aus dem Blog verschwunden.Seit dem habe ich zweimal versucht Fee und Uli zu kontaktieren, leider aber keine Antwort erhalten. Ein weiterer Kommentar wurde gelöscht.
Ähnliche Fehler wurden in der populärwissenschaftlichen Berichterstattung über das Platypus-Genom-Paper haufenweise gemacht und un z.B. von T.Ryan Gregory sehr gut diskutiert.
Meine Meinung dazu:
Im deutschsprachigen Raum fehlt bislang eine Diskussionskultur auf Wissenschaftsblogs. Researchblogging ist sicher ein guter Ansatz dies zu ändern. Kritik an Blogposts muss auch in ungeschminkter Form erlaubt sein (dieser Post ist übrigens genauso grottenschlecht, und wird von Argent23 "mit Samthandschuhen" kritisiert). Der Name des Blogs (Science-meets-society; science communication) ist im Zusammenhang mit der Löschung unerwünschter Kommentare geradezu lächerlich.
Ich bin ein großer Fan guter Vereinfachungen von komplexen Zusammenhängen in populärwissenschaftlichen Texten zur Evolutionsbiologie und allgemein von Wissenschaftsjournalismus. Allerdings braucht man um den betreffenden Sprachgebrauch zu beherrschen eine fundierte Kenntnis des Fachgebiets und einige Erfahrung (wegen letzterem lesen sich manche meiner Posts vielleicht auch etwas kryptisch). Gerade die Vereinfachung macht die Sache eben nicht leichter.
Zum thematischen Ursprung des Disputs noch soviel: Wer sich weiter informieren möchte kann dies auf deutschen Blogs noch nicht (falls doch bin ich dankbar für nen Link). Ich werd mich drum kümmern sobald ich etwas mehr Zeit hab! Über Fallstricke im Verständnis von Evolutionsbiologie kann man sich solange super (besser als ich das wahrscheinlich jemals hinbekommen werde) bei T. Tyan Gregory informieren.
Samstag, 20. September 2008
Powerful statistics
Jeder, der schonmal gedacht hat er hätte in einer Präsentation chronologische Daten optimal visualisiert sollte sich das hier mal anschauen...
Hans Rosling bei einem TED-Vortrag
Wie geil ist da eigentlich? Die Idee dahinter nennt sich Gapminder und ist als Google Gadget verfügbar. Jetzt brauch ich nur noch Wurm-Daten, die so ne Darstellung erlauben :-).
Hans Rosling bei einem TED-Vortrag
Wie geil ist da eigentlich? Die Idee dahinter nennt sich Gapminder und ist als Google Gadget verfügbar. Jetzt brauch ich nur noch Wurm-Daten, die so ne Darstellung erlauben :-).
Freitag, 19. September 2008
Montag, 8. September 2008
Würmer in Paris: Compatibility polymorphism in snail/trematode interactions
Die Interaktion von Trematoden und Schnecken beruht (nach der aktuellen Hypothese) darauf, dass das sich zur Muttersporocyste entwickelnde Miracidium des Parasiten den Wirt entweder aktiv in seiner Immunantwort beeinflusst, oder aber durch ein Mimikry von Wirtsepitopen vom Immunsystem unerkannt bleibt. Letzteres ist bei B. glabrata und S. mansoni der Fall.
Während bei einer Beeinflussung/Unterdrückung des Immunsystems in anderen Wirt/Parasit-Systemen generell von anfälligen und resistenten Wirten gesprochen werden kann, ist die Interaktion beim Mimikry von Wirtsepitopen komplizierter:
Die selbe Parasiten-Linie ist unterschiedlich kompatibel mit künstlich auf Resistenz oder Anfälligkeit selektierten Schnecken-Linien (Soweit wäre das natürlich auch durch bloße Resistenz der Schnecke erklärbar). Werden aber anderer Schnecken-Linien mit einer anderen Parasiten-Linie selektiert ist die in dieser künstlichen Co-Evolution resistente Schneckenlinie anfällig für die Parasiten-Linie aus der ersten Selektion. Ein schönes Schaubild dazu (Fig.3) kann man sich in diesem Paper anschauen, wenn man Zugriff hat.
Die Versuche der Gruppe zur Identifizierung der molekularen Grundlage dieser Interaktion wurden nun mit einem zu einer brasilianischen Schnecken-Linie kompatiblen Stamm von S. mansoni und einem inkompatiblen Stamm durchgeführt (Zur Vermehrung des inkompatiblen Stamms stand eine andere Schneckenlinie zur Verfügung).
Verwendet wurde ein proteomischer Ansatz (2D Gele) und es konnten sogenannte Schistosoma mansoni polymorphic mucin-like proteins (Sm PoMuc) als Hauptverdächtige identifiziert werden.
Mucine oder Mucin-ähnliche Proteine sind im Schleim (Mucus) vieler Organismen enthalten. Solcher Mucus wird oft von als Reaktion auf eine Infektion produziert, und kann dazu dienen Parasiten zu bekämpfen. Andererseits sekretieren Parasiten aber auch Mucin-ähnliche Moleküle um die Wirtsabwehr zu täuschen.
In Sm PoMucs werden fast ausschließlich in den in der Schnecke parasitierenden Stadien exprimiert. In den Miracidien werden sie in sogenannten Apicaldrüsen produziert und sekretiert. Möglicherweise kann sich der Parasit so in einem küstlichen Nebelschleier verbergen (im Deutschen klingt das schon fast poetisch; anders als das Englische "covered by a smoke screen").
Die Struktur der Sm PoMucs erwies sich als höchst polymorph. Gemeinsam haben alle Transkripte ein Signalpeptid (in Übereinstimmung mit der extrazellulären Lokalisation) gefolgt von einer Region bestehend aus einer variablen Anzahl von (3 leicht unterschiedlichen; r1, r1' und r2) 9-Aminosäuren-Repeats gefolgt von 3 leicht unterschiedlichen C-terminalen Regionen (1, 2, 3).
Eine unterschiedliche Kombination dieser Elemente kennzeichnet drei Gruppen von Sm PoMucs (die erste Gruppe und die zweite Gruppe haben nur r2 und unterschiedliche C-terminale Regionen 1 und 2; die dritte Gruppe hat R1' und R1 gefolgt von der C-terminalen Region 3).
Eine vierte Gruppe wiest eine wahrscheinlich durch alternatives Splicen noch stärker abweichende Sequenz auf.
Im Kompatiblen und inkompatiblen Parasiten-Stamm unterscheidet sich in allen drei Gruppen die Anzahl der Repeats. Codiert werden die drei Gruppen von jewels einem Gen. Die unterschiedliche Anzahl der Repeats entsteht ebenfalls durch alternatives splicen.
Besonders von Bedeutung ist weiterhin, das die Repeats Serin-, Threonin- und Prolin-reich sind, was als Zeichen für eine posttranslationale Glycolsylierung gilt.
So könnten möglicherweise speziell die unterschiedlich angefügten Zucker für die unterschiedlichen Eigenschaften der Moleküle in der Wirts-Parasit-Interaktion verantwortlich sein.
Für mich war der Vortrag deshalb so interessant, da er mir die Beschränkungen der in meinem Projekt verwendeten transkriptomischen Methoden vor Augen geführt hat:
Allein das Assembley der Repeats wäre aus 454-Pyrosequenz-Daten (bei der bisherigen read-Länge von 250 Basen) wahrscheinlich sehr schwierig bis unmöglich. Und selbst mit sehr viel längeren reads bräuchte man eine extrem hohe Coverage der betreffenden Regionen um ein solches Maß an Polymorphismus aufdecken zu können.
Außerdem interessant (und etwas beunruhigend, wenn man die Arbeit mit Proteinen nicht besonders mag) ist die Tatsache, dass ein wesentlicher Teil des Polymorphismus durch posttranslationale Modifikation entstehen kann. Generell neu und überraschend ist das natürlich nicht unbedingt, allerdings könnte es besonders im Co-Evolutions-Context eine gewaltige Rolle spielen.
Gerade in der Interaktion mit mehreren sympatrischen Wirtsarten könnten solche Mechanismen zu einer Evolution von Plastizität führen. Speziell diese Plastizität könnte als Gegenspieler von adaptiven Prozessen sympatrische, ökologische Artbildung von den an unterschiedliche Wirtsorganismen angepassten Parasiten-Stämmen verhindern.
_______________________________________________________________________________________________________________________________________
E ROGER, B GOURBAL, C GRUNAU, R PIERCE, R GALINIER, G MITTA (2008). Expression analysis of highly polymorphic mucin proteins (Sm PoMuc) from the parasite Schistosoma mansoni☆ Molecular and Biochemical Parasitology, 157 (2), 217-227 DOI: 10.1016/j.molbiopara.2007.11.015
Samstag, 6. September 2008
Würmer in Paris: Ist DNA-barcoding unfair?
Ein Großteil der Diskussion nach dem DNA-barcoding Symposium auf dem EMOP hatte anstatt der technischen Schwierigkeiten [1] leider eine andere Grundlage:
3. R. Guerrero stellte die Schwierigkeiten der Forscher aus "Schwellenländern" sich umfangreiche Sequenzierungen finanziell leisten zu können in den Vordergrund:
Dadurch würden gerade in diesen Ländern, in denen die Biodiversität am größten ist keine Fortschritte erzielt, während in den entwickelten Ländern, mit fast vollständig beschriebener Diversität nach kryptischen Arten gesucht wird.
Ganz von der Hand zu weißen sind diese Einwände wahrscheinlich nicht, da selbst wenn Sequenzierungen immer günstiger werden, das Anlegen von großen Datenbanken, die Morphologie (Museumsexemplare, Bilder, Videos), ökologische Parameter (Verbreitung) mit den Sequenzdaten verknüpfen immer noch teuer bleibt.
Andererseits: Kein Doktorrand wird sich dagegen sträuben im Rahmen seiner Dissertation in die Tropen zu reisen um umfangreiche Proben zu nehmen, die dann daheim sequenziert werden. Und auch der durchschnittliche Arbeitsgruppenleiter auf seinem speziellen Gebiet wird (sobald Hochdurchsatzsequenzierung mal wirklich nichts mehr kostet und weit verbreitet ist) den Vorteil und das Prestige erkennen den die Entdeckung hunderter "möglicher Arten" bietet.
[1] Davon gibt es (scheinbar?) genügend: Alleine F1000 listet 3 Paper, die von Problemen berichten (in Fällen, wo die intraspezifische Varianz die interspezifische überlappt) und 3 Paper die Erfolge beschreiben.
Zurzeit gibt es außerdem ein viel diskutiertes Paper über Probleme durch COI-Pseudogene im Kern...
...das ganze ist schwer zu beurteilen und natürlich spielen dabei die persönlichen Arbeitsweisen eine Rolle. Ein deutscher Evolutionsökolge meinte scherzhaft zu einem Kollegen, als ich ihn darauf ansprach dass ich gerne alle Aalparasiten barcoden würde: "Vorsicht, der will uns wegrationalisieren: Dann schmeisst er nur noch alles in nen Mixer und zählt Sequenzen..."
3. R. Guerrero stellte die Schwierigkeiten der Forscher aus "Schwellenländern" sich umfangreiche Sequenzierungen finanziell leisten zu können in den Vordergrund:
Dadurch würden gerade in diesen Ländern, in denen die Biodiversität am größten ist keine Fortschritte erzielt, während in den entwickelten Ländern, mit fast vollständig beschriebener Diversität nach kryptischen Arten gesucht wird.
Ganz von der Hand zu weißen sind diese Einwände wahrscheinlich nicht, da selbst wenn Sequenzierungen immer günstiger werden, das Anlegen von großen Datenbanken, die Morphologie (Museumsexemplare, Bilder, Videos), ökologische Parameter (Verbreitung) mit den Sequenzdaten verknüpfen immer noch teuer bleibt.
Andererseits: Kein Doktorrand wird sich dagegen sträuben im Rahmen seiner Dissertation in die Tropen zu reisen um umfangreiche Proben zu nehmen, die dann daheim sequenziert werden. Und auch der durchschnittliche Arbeitsgruppenleiter auf seinem speziellen Gebiet wird (sobald Hochdurchsatzsequenzierung mal wirklich nichts mehr kostet und weit verbreitet ist) den Vorteil und das Prestige erkennen den die Entdeckung hunderter "möglicher Arten" bietet.
[1] Davon gibt es (scheinbar?) genügend: Alleine F1000 listet 3 Paper, die von Problemen berichten (in Fällen, wo die intraspezifische Varianz die interspezifische überlappt) und 3 Paper die Erfolge beschreiben.
Zurzeit gibt es außerdem ein viel diskutiertes Paper über Probleme durch COI-Pseudogene im Kern...
...das ganze ist schwer zu beurteilen und natürlich spielen dabei die persönlichen Arbeitsweisen eine Rolle. Ein deutscher Evolutionsökolge meinte scherzhaft zu einem Kollegen, als ich ihn darauf ansprach dass ich gerne alle Aalparasiten barcoden würde: "Vorsicht, der will uns wegrationalisieren: Dann schmeisst er nur noch alles in nen Mixer und zählt Sequenzen..."
Würmer in Paris: Acoel Flatworms
2. Tim Littlewood brachte den besten Witz während eines Vortrags und schloss gleichzeitig eine Lücke in meinem Wissen über Phylogenie der Protostomier:
Er erwähnte, dass sehr viele lateinische Taxa-bezeichnungen in verschiedenen Sprachen völlig unterschiedlich ausgesprochen würden. So würde "Acoel" schon im deutschen und englischen etwas unterschiedlich ausgesprochen, die Franzosen sprächen es sogar "ashole" aus. Originalzitat: "but dont be a ashole (acoel) try to understand each other"...
Was hat es nun mit diesen (nicht-parasitischen) Würmern auf sich?
Wie man als halbwegs gebildeter Biologie (und erst recht als Zoologe im weitesten Sinne) wissen sollte hat die Phylogenie der Tiere in den letzen zehn Jahren [1] eine Revision erfahren [2]:
Dabei gibt es zwei große Kladen innerhalb der bilateralsymmetrischen Tiere, die Protostomier und die Deuterostomier (zu denen wir Vertebraten gehören).
Die Protostomier enthalten wiederum zwei Kladen: Eine die alle Tiere mit "sich häutenden" (moulting) Larvenstadien (Nematoden, Cheliceraten, etc.) umfasst und Ectysozoa genannt wird und eine zweite die Lophotrochozoa genannt wird.
Der Name Lophotrochozoa ist eine Neuschöpfung aus den Namen der beiden alten Kladen Trochozoa und Lophophorata.
Diese Lophotrochozoa enthalten neben den prominenten Mollusken und Anneliden auch die Platyhelminthen (hauptsächlich Trematoden und Cestoden für die Parasitologen). Dise alte Klade erwies sich aber eben auch als polyphyletisch und die "Acoel Flatworms" mussten ausgegliedert und an eine basale Position [3] als Schwestergruppe der übrigen Bilateria [4] gestellt werden.
Quellen im Text verlinkt!
Wobei ich jeweils zu der genererellen Revision [1+2] und den "Acoel Flatworms" [3+4] DAS klassische und ein neues Paper mit einer phylogenomischen Analyse verlinkt hab. (Phylogenomik bezieht sich hier auf die Analyse mehrerer (vieler) Genen aus großen EST-Datensätzen, nicht auf Jonathan Eisens Definition.)
Er erwähnte, dass sehr viele lateinische Taxa-bezeichnungen in verschiedenen Sprachen völlig unterschiedlich ausgesprochen würden. So würde "Acoel" schon im deutschen und englischen etwas unterschiedlich ausgesprochen, die Franzosen sprächen es sogar "ashole" aus. Originalzitat: "but dont be a ashole (acoel) try to understand each other"...
Was hat es nun mit diesen (nicht-parasitischen) Würmern auf sich?
Wie man als halbwegs gebildeter Biologie (und erst recht als Zoologe im weitesten Sinne) wissen sollte hat die Phylogenie der Tiere in den letzen zehn Jahren [1] eine Revision erfahren [2]:
Dabei gibt es zwei große Kladen innerhalb der bilateralsymmetrischen Tiere, die Protostomier und die Deuterostomier (zu denen wir Vertebraten gehören).
Die Protostomier enthalten wiederum zwei Kladen: Eine die alle Tiere mit "sich häutenden" (moulting) Larvenstadien (Nematoden, Cheliceraten, etc.) umfasst und Ectysozoa genannt wird und eine zweite die Lophotrochozoa genannt wird.
Der Name Lophotrochozoa ist eine Neuschöpfung aus den Namen der beiden alten Kladen Trochozoa und Lophophorata.
Diese Lophotrochozoa enthalten neben den prominenten Mollusken und Anneliden auch die Platyhelminthen (hauptsächlich Trematoden und Cestoden für die Parasitologen). Dise alte Klade erwies sich aber eben auch als polyphyletisch und die "Acoel Flatworms" mussten ausgegliedert und an eine basale Position [3] als Schwestergruppe der übrigen Bilateria [4] gestellt werden.
Quellen im Text verlinkt!
Wobei ich jeweils zu der genererellen Revision [1+2] und den "Acoel Flatworms" [3+4] DAS klassische und ein neues Paper mit einer phylogenomischen Analyse verlinkt hab. (Phylogenomik bezieht sich hier auf die Analyse mehrerer (vieler) Genen aus großen EST-Datensätzen, nicht auf Jonathan Eisens Definition.)
Würmer in Paris: Orientation within host tissues
Ich fange heute an einen längeren Post über den in meinen Augen interessantesten Vortrag der EMOP zu schreiben, dazu wollen aber (mangels Mitschrieb) noch einige Paper gelesen werden.
Zuächst aber kurze Zusammenfassungen einiger anderer Vorträge und Diskussionen.
1. Wilfried Haas: ORIENTATION WITHIN HOST TISSUES: MECHANISMS OF PARASITE NAVIGATION
Im Labor von Prof. Haas in Erlangen konnte in den letzen Jahren durch in vivo Experimenten eine Wanderung von Trematoden (und mit in geringerem Umfang auch von Nematoden) entlang von chemischen Gradienten demonstriert werden. Alle getesteten Helminthen außer der Nematode Necator americanus wanderten dabei in Agar entlang von Gradienten des Wirtsplasmas. Die Gruppe demonstrierte dann durch eine Auftrennung des Wirtsplasmas und umfangreiche Tests, dass für die untersuchten Trematoden Arginin, Glucose und Mannose anziehend wirken, Glucosamin abstoßend. Der zweite Nematode Ancylostoma duodenale wanderte in Richtung anorganischer Ionen. Diese Ergebnisse machen durchaus evolutionsökologischen Sinn, da die Metacercarien von Trematoden, sobald sie in den Endwirt eingedrungen sind, empfindlich gegenüber Sauerstoff werden, und so sicherstellen müssen, dass sie in tiefere Gewebeschichten vordringen (wo Arginin , Glucose und Mannose vorkommen) und eine erneutes durchstoßen der Wirtshaut (in der Glucosamin vorkommt) vermeiden.
Ich kann hier trotzdem nicht auf die Veröffentlichungen der Gruppe verlinken, da die Ergebnisse der vorherrschenden Meinung widersprechen, dass Parasiten (inkl. Tremtoden) lediglich genetisch programmierten Wanderruten in der Leibeshöhle des Wirtes folgen.
So wurden laut Prof. Haas bisher alle Veröffentlichungen dieser Arbeiten vom wichtigsten Vertreter der vorherrschenden Lehrmeinung Prof. Michael V. K. Sukhdeo geblockt. Die Gründe dafür sind, dass ein in vivo System nicht als Grundlage für die Revision einer solch etablierten Lehrnmeinung anerkannt wird. Die Gruppe um Prof. Haas war die erste Gruppe weltweit der überhaupt die Demonstration des beschriebenen Phänomens gelang, da die Einstellung der Gradienten von sehr geringen Konzentrationen offenbar in sehr vielen Versuchen anderer Gruppen zuvor misslang.
Zuächst aber kurze Zusammenfassungen einiger anderer Vorträge und Diskussionen.
1. Wilfried Haas: ORIENTATION WITHIN HOST TISSUES: MECHANISMS OF PARASITE NAVIGATION
Im Labor von Prof. Haas in Erlangen konnte in den letzen Jahren durch in vivo Experimenten eine Wanderung von Trematoden (und mit in geringerem Umfang auch von Nematoden) entlang von chemischen Gradienten demonstriert werden. Alle getesteten Helminthen außer der Nematode Necator americanus wanderten dabei in Agar entlang von Gradienten des Wirtsplasmas. Die Gruppe demonstrierte dann durch eine Auftrennung des Wirtsplasmas und umfangreiche Tests, dass für die untersuchten Trematoden Arginin, Glucose und Mannose anziehend wirken, Glucosamin abstoßend. Der zweite Nematode Ancylostoma duodenale wanderte in Richtung anorganischer Ionen. Diese Ergebnisse machen durchaus evolutionsökologischen Sinn, da die Metacercarien von Trematoden, sobald sie in den Endwirt eingedrungen sind, empfindlich gegenüber Sauerstoff werden, und so sicherstellen müssen, dass sie in tiefere Gewebeschichten vordringen (wo Arginin , Glucose und Mannose vorkommen) und eine erneutes durchstoßen der Wirtshaut (in der Glucosamin vorkommt) vermeiden.
Ich kann hier trotzdem nicht auf die Veröffentlichungen der Gruppe verlinken, da die Ergebnisse der vorherrschenden Meinung widersprechen, dass Parasiten (inkl. Tremtoden) lediglich genetisch programmierten Wanderruten in der Leibeshöhle des Wirtes folgen.
So wurden laut Prof. Haas bisher alle Veröffentlichungen dieser Arbeiten vom wichtigsten Vertreter der vorherrschenden Lehrmeinung Prof. Michael V. K. Sukhdeo geblockt. Die Gründe dafür sind, dass ein in vivo System nicht als Grundlage für die Revision einer solch etablierten Lehrnmeinung anerkannt wird. Die Gruppe um Prof. Haas war die erste Gruppe weltweit der überhaupt die Demonstration des beschriebenen Phänomens gelang, da die Einstellung der Gradienten von sehr geringen Konzentrationen offenbar in sehr vielen Versuchen anderer Gruppen zuvor misslang.
Freitag, 5. September 2008
A moveable feast
Paris war der Hammer! Über die wissenschaftlichen Anregungen werde ich in Kürze schreiben!

Das war das Tagungsgebäude...
Nach den ersten vier Paris-Tagen hab ich mir allerdings noch ein paar Tage Urlaub gegönnt.

Das war das Tagungsgebäude...
Nach den ersten vier Paris-Tagen hab ich mir allerdings noch ein paar Tage Urlaub gegönnt.
Abonnieren
Kommentare (Atom)
