Leider habe ich es nicht geschafft einen zusammenfassenden Paper-Diskussion Post zur neutralen Theorie vor meinem Urlaub ferstigzustellen.
Schade... ab 17.01.2009 geht's hier weiter. Guten Rutsch!
Freitag, 26. Dezember 2008
Sonntag, 30. November 2008
Google und die deutsche Evolutionsbiolgie
Zu meinem großen Erstaunen konnte ich eben feststellen, dass ich für den Suchbegriff
den Top-Hit von insgesamt 4010 Treffern auf Google habe. Die äquivalente Englischsprachige Suche ergibt 31300 Treffer mit einem Wikipedia-Eintrag an erster Stelle.
Sehr beunruhigend ist, dass kreationistische Texte wie das deutschen Intelligent-Design Lehrbuch "Evolution, ein kritisches Lehrbuch" von Reinhard Junker und Siegfried Scherer und eine positive Rezension zu diesem, zwei der ersten zehn Plätze einnehmen. Ein dritter Platz unter den ersten zehn bei dieser Suche geht an www.kritische-naturgeschichte.de, eine wissenschaftsfeindliche deutsche Seite, die - scheinbar ideologisch unabhängig - versucht die wissenschaftliche Methode als unanwendbar in der Biologie darzustellen.
Nach der ersten Freude über die Google-Suche, bin mir meiner Verantwortung bewusst, die "Fackel der Aufklärung" im deutschsprachigen Raum hochzuhalten.
Die nahezu neutrale Theorie der molekularen Evolution
den Top-Hit von insgesamt 4010 Treffern auf Google habe. Die äquivalente Englischsprachige Suche ergibt 31300 Treffer mit einem Wikipedia-Eintrag an erster Stelle.
Sehr beunruhigend ist, dass kreationistische Texte wie das deutschen Intelligent-Design Lehrbuch "Evolution, ein kritisches Lehrbuch" von Reinhard Junker und Siegfried Scherer und eine positive Rezension zu diesem, zwei der ersten zehn Plätze einnehmen. Ein dritter Platz unter den ersten zehn bei dieser Suche geht an www.kritische-naturgeschichte.de, eine wissenschaftsfeindliche deutsche Seite, die - scheinbar ideologisch unabhängig - versucht die wissenschaftliche Methode als unanwendbar in der Biologie darzustellen.
Nach der ersten Freude über die Google-Suche, bin mir meiner Verantwortung bewusst, die "Fackel der Aufklärung" im deutschsprachigen Raum hochzuhalten.
Populationsgenetik Serie: Organisatorisches
Ich werde hier in den nächsten Wochen und Monaten hauptsächlich über Populationsgenetik schreiben. Die betreffenden Posts tragen das entsprechende Label. Meine Motivation und meine generellen Hauptquellen dazu beschreibe ich in diesem Eröffnungspost.
Die Posts werden sich in vier Kategorien gliedern:
Gleiches gilt für Grafiken, die in geschweiften Klammern beschriftet werden z.B. {2}.
Grafiken werden von mir selbst mit Hilfe von R erstellt. Am Ende von Post, die mehr oder weniger aufwändige Plots enthalten wird der Leser einen Link zu einem funktionierenden R-script finden. Der Text aus diesen Google-Docs kann in einen Texteditor kopiert, modifiziert und danach in R zum erstellen eigener Grafiken benutzt werden.
Ich möchte dem interessierten Leser so die Möglichkeit geben selbst etwas mit den Formeln zu spielen und gleichzeitig -mit mir zusammen- einen Einstieg in R zu finden.
Ein Einführung in Populationsgenetik mit Hilfe von R - Eine Einführung in R mit Hilfe von Populationsgenetik.
Viel Spass!
Die Populationsgenetik Serie zieht um! Da die Beiträge untereinander sehr stark verknüpft sind und die Serie andernorts fortgesetzt wird, werde ich alle Posts der Serie in den nächsten Tagen auf den neuen Blog Alles was lebt bringen.
Die Posts werden sich in vier Kategorien gliedern:
- Grundlagen [Beispiel]
- Kernkonzepte, Modelle und Theorien [Beispiel]
- Paperdiskussion [Beispiel]
- Nebensächliches [Beispiel]
Gleiches gilt für Grafiken, die in geschweiften Klammern beschriftet werden z.B. {2}.
Grafiken werden von mir selbst mit Hilfe von R erstellt. Am Ende von Post, die mehr oder weniger aufwändige Plots enthalten wird der Leser einen Link zu einem funktionierenden R-script finden. Der Text aus diesen Google-Docs kann in einen Texteditor kopiert, modifiziert und danach in R zum erstellen eigener Grafiken benutzt werden.
Ich möchte dem interessierten Leser so die Möglichkeit geben selbst etwas mit den Formeln zu spielen und gleichzeitig -mit mir zusammen- einen Einstieg in R zu finden.
Ein Einführung in Populationsgenetik mit Hilfe von R - Eine Einführung in R mit Hilfe von Populationsgenetik.
Viel Spass!
Die Populationsgenetik Serie zieht um! Da die Beiträge untereinander sehr stark verknüpft sind und die Serie andernorts fortgesetzt wird, werde ich alle Posts der Serie in den nächsten Tagen auf den neuen Blog Alles was lebt bringen.
Die nahezu neutrale Theorie der molekularen Evolution
Eine elegante Erweiterung der neutralen Theorie haben Kimura und Ohta in den späten 1980ern entwickelt. Sie haben folgende die Gleichung für die Fixierungswahrscheinlichkeit von Mutationen, die schwach selektiert werden gefunden:
 [9]
[9]
s ist dabei der sogenannte Selektionskoeffizient relativ zur durchschnittlichen Fitness. In dem zugrunde liegenden Modell hat das in der Population bereits vorhandene Allel die Fitness 1, Homozygote für die neue Mutation haben die Fitness 1+s, Heterozygote 1+s/2.
Folgendes Schaubild zeigt den Einfluss der Populationsgröße auf die Effektivität der Selektion. Der Selektionskoeffizient s ist dabei von -0.02 (2% schlechtere Fitness/Fortpflanzungswahrscheinlichkeit der Homozygoten für die Mutation, 1% der Heterozygoten ) bis 0,01 (1% besseres Abschneiden der Homozygoten, 0,5 der Heterozygoten) aufgetragen.

{2}
Deutlich wird, dass in grösseren Populationen die Selektion wirksamer ist. Nes sollte um die Formel interessant zu machen in der Nähe von 1 liegen, der von mir angenommene Selektionskoeffizient von -0.02 bis 0.01ist vergleichsweise groß und daher gibt die Formel für eher kleine Werte von N interessante Graphen. In realistischeren Situationen wird die Formel wohl eher bei um einige Zehnerpotenzen größeren Populationen angewandt deren Selektionskoeffizient um einige Zehnerpotenzen kleiner sind.
Schön zu sehen ist auch dass die Formel "im Limit" (s=0) die gleichen Werte gibt wie Formel [1].
Die R-Befehle gibt es hier als googledoc. Einfach in eine Textdatei einfügen. Das ganze als deinscript.R speichern. In R erzeugt das script mit source("deinpfad/deinscript.R") den Plot dieses Posts mit dem Namen nearlyneutralauto.jpg in dem Ordner in dem du R gestartet hast.
[9]s ist dabei der sogenannte Selektionskoeffizient relativ zur durchschnittlichen Fitness. In dem zugrunde liegenden Modell hat das in der Population bereits vorhandene Allel die Fitness 1, Homozygote für die neue Mutation haben die Fitness 1+s, Heterozygote 1+s/2.
Folgendes Schaubild zeigt den Einfluss der Populationsgröße auf die Effektivität der Selektion. Der Selektionskoeffizient s ist dabei von -0.02 (2% schlechtere Fitness/Fortpflanzungswahrscheinlichkeit der Homozygoten für die Mutation, 1% der Heterozygoten ) bis 0,01 (1% besseres Abschneiden der Homozygoten, 0,5 der Heterozygoten) aufgetragen.
{2}
Deutlich wird, dass in grösseren Populationen die Selektion wirksamer ist. Nes sollte um die Formel interessant zu machen in der Nähe von 1 liegen, der von mir angenommene Selektionskoeffizient von -0.02 bis 0.01ist vergleichsweise groß und daher gibt die Formel für eher kleine Werte von N interessante Graphen. In realistischeren Situationen wird die Formel wohl eher bei um einige Zehnerpotenzen größeren Populationen angewandt deren Selektionskoeffizient um einige Zehnerpotenzen kleiner sind.
Schön zu sehen ist auch dass die Formel "im Limit" (s=0) die gleichen Werte gibt wie Formel [1].
Die R-Befehle gibt es hier als googledoc. Einfach in eine Textdatei einfügen. Das ganze als deinscript.R speichern. In R erzeugt das script mit source("deinpfad/deinscript.R") den Plot dieses Posts mit dem Namen nearlyneutralauto.jpg in dem Ordner in dem du R gestartet hast.
Die neutrale Theorie der molekularen Evolution, Teil 2
Vielleicht ist es jemandem aufgefallen: Die Teilaspekte der neutrale Theorie, die ich im ersten Post vorgestellt hatte erlauben nicht unbedingt viele Voraussagen und wären zu Kimuras Zeit, vor Entwicklung der DNA-Sequenzierung, in dieser Form untestbar gewesen.
Wie ich bereits angedeutet habe hat die Theorie auch einen mathematisch etwas schwierigen Teil: Formel, die von Kimura aus Diffusionsgleichungen ableitete, da Mutationen ähnlich diesem physikalischen Prinzip in die Population "diffundieren". Für die durchschnittliche Zeit zwischen dem entstehen der Mutation und ihrer Fixierung konnte er 4Ne Generationen ermitteln.
Mutationen, die zum verschwinden verurteilt sind tun dies dagegen im Durchschnitt innerhalb von
 [6]
[6]
Generationen.
Mutationen die verloren gehen tun dies als in wesentlich kürzerer Zeit, als solche die fixiert werden.
Nehmen wir nen weiter ein Modell mit unendlich vielen verschiedenen möglichen Allelen an, gibt
 [7]
[7]
die erwartete Homozygotie unter Neutralität in einem Gleichgewichtszustand von Mutation und Verlust der Mutationen durch Drift. u ist dabei die wieder die neutrale Mutationsrate. Die Formel hat eine schöne Herleitung auf die ich in späteren Posts zurückkommen werde, da dafür noch weitere Konzepte erklärt werden müssen.
Homozygotie beschreit den Zustand eines Locus (Genort) an dem nur eine Allel in der Population vorhanden ist. Dieser "Zustand" ist heute erkennbar indem man den betreffenden Locus für genügend Individuen der Population sequenziert, diese Technik war allerdings erst seit den achtziger Jahren verfügbar und auch bis in die neunziger für eine breite Anwendung noch zu teuer und arbeitsaufwändig.
Man hat nun bereits zu Kimuras Zeit festgestellt, dass diese Voraussagen über Homozygotität für anhand von Allozym -Polymorphismen gewonnenen Daten nicht immer mit der Realität übereinstimmen. Allozyme waren in der Zeit nach der Entwicklung der Populationsgenetik lange Zeit das einzige Werkzeug um Einblicke in die Genetik jenseits von morphologischen, diskreten Merkmalen, wie sie Mendel benutzt hatte, zu erlangen. Ich sollte ihnen einen eigenen Post widmen...
Für die meisten Daten wird aus historischen Gründen, die wir noch kennen lernen werden, eher die die Heterozygotie als die Homozygotie angegeben. Da jeder Lokus entweder im einen oder im anderen Zustand vorliegt, ist der Zusammenhang zwischen beiden Messwerten aber ein einfacher: Die Heterozygotie (H) ist 1- die Homozygotie. Deshalb ist die einfache Umformung von Formel [7]:
 [8]
[8]
Im nächsten Post werde ich zunächst auf die nahezu neutrale Version der Theorie eingehen, dann sind die Grundlagen vorhanden um einige Veröffentlichungen -auch aktuelle- zu besprechen.
Wie ich bereits angedeutet habe hat die Theorie auch einen mathematisch etwas schwierigen Teil: Formel, die von Kimura aus Diffusionsgleichungen ableitete, da Mutationen ähnlich diesem physikalischen Prinzip in die Population "diffundieren". Für die durchschnittliche Zeit zwischen dem entstehen der Mutation und ihrer Fixierung konnte er 4Ne Generationen ermitteln.
Mutationen, die zum verschwinden verurteilt sind tun dies dagegen im Durchschnitt innerhalb von
[6]Generationen.
Mutationen die verloren gehen tun dies als in wesentlich kürzerer Zeit, als solche die fixiert werden.
Nehmen wir nen weiter ein Modell mit unendlich vielen verschiedenen möglichen Allelen an, gibt
[7]die erwartete Homozygotie unter Neutralität in einem Gleichgewichtszustand von Mutation und Verlust der Mutationen durch Drift. u ist dabei die wieder die neutrale Mutationsrate. Die Formel hat eine schöne Herleitung auf die ich in späteren Posts zurückkommen werde, da dafür noch weitere Konzepte erklärt werden müssen.
Homozygotie beschreit den Zustand eines Locus (Genort) an dem nur eine Allel in der Population vorhanden ist. Dieser "Zustand" ist heute erkennbar indem man den betreffenden Locus für genügend Individuen der Population sequenziert, diese Technik war allerdings erst seit den achtziger Jahren verfügbar und auch bis in die neunziger für eine breite Anwendung noch zu teuer und arbeitsaufwändig.
Man hat nun bereits zu Kimuras Zeit festgestellt, dass diese Voraussagen über Homozygotität für anhand von Allozym -Polymorphismen gewonnenen Daten nicht immer mit der Realität übereinstimmen. Allozyme waren in der Zeit nach der Entwicklung der Populationsgenetik lange Zeit das einzige Werkzeug um Einblicke in die Genetik jenseits von morphologischen, diskreten Merkmalen, wie sie Mendel benutzt hatte, zu erlangen. Ich sollte ihnen einen eigenen Post widmen...
Für die meisten Daten wird aus historischen Gründen, die wir noch kennen lernen werden, eher die die Heterozygotie als die Homozygotie angegeben. Da jeder Lokus entweder im einen oder im anderen Zustand vorliegt, ist der Zusammenhang zwischen beiden Messwerten aber ein einfacher: Die Heterozygotie (H) ist 1- die Homozygotie. Deshalb ist die einfache Umformung von Formel [7]:
[8]Im nächsten Post werde ich zunächst auf die nahezu neutrale Version der Theorie eingehen, dann sind die Grundlagen vorhanden um einige Veröffentlichungen -auch aktuelle- zu besprechen.
Samstag, 29. November 2008
Grundlagen: Effektive Populationsgröße
Wie wir im ersten Post über die neutrale Theorie gesehen haben spielt bei Zufallsprozessen, wie genetischem Drift (der zufälligen Fixierung bestimmter Allele) , die Populationsgröße eine Rolle. Wir haben für diesen ersten Teil dieser Theorie lediglich die aktuelle Populationsgroesse N betrachtet, diese kann man "einfach" durch "zählen" der betreffenden Individuen der Population bestimmen. Dies funktioniert leider nur auf Kosten mehrerer Voraussetzungen, wie gleichbleibender Populationsgröße und zufälliger Paarung.
Wollen wir unsere Modelle nun aber auf realistischere Systeme anwenden, brauchen wir das Konzept der effektiven Populationsgröße(Ne).
Viele Population haben beispielsweise eine ungleiche Anzahl sich fortpflanzender Männchen und Weibchen. Dies ist bei starker männlicher Konkurrenz um die Weibchen der Fall, wo sich in jeder Generation nur ein Bruchteil der Männchen fortpflanzen.
In diesem Fall ist
 [3]
[3]
Wobei Nm die Anzahl der sich fortpflanzenden Männchen, Nf die Anzahl der sich fortpflanzenden Weibchen ist.
Spielt man etwas mit dieser Formel, wir beim Einsetzen von Werten sehr schnell deutlich, dass wenn Nm sehr viel größer als Nf der Wert für Ne eher in der Nähe des kleinen Wertes liegt. Dies macht Sinn, da durch die wenigen sich paarenden Männchen in jeder neuen Generation eine Art Flaschenhals ensteht: Die Hälfte der autosomal weitergegebenen Allele wird durch einen kleinen Bruchteil der Individuen weitergegeben.
Ähnliches gilt aus den gleichen Gründen, wenn sich einzelne Individuen -unabhängig vom Geschlecht- sehr unterschiedliche Nachkommenzahlen haben, dann ist
 [4]
[4]
wobei Vk die Varianz in der Nachkommenzahl ist. Da bei gleichbleibender Populationsgröße (bisher immer noch eine Ausgangsannahme) durchschnittlich zwei Nachkommen pro Elternteil entstehen, ist die Nachkommenanzahl bei zufälliger Fortpflanzung Poisson-verteilt mit einem Mittelwert und einer Varianz von 2. Größere Varianzen lassen in obiger Formel Ne kleiner als N werden.
Doch was passiert wenn sich die Populationsgröße über die Zeit ändert? Ganz einfach
 [5]
[5]
d.h. Ne ist das harmonische Mittel der Populationsgrößen in n Generationen.
Ähnliche Gleichungen für die effektiven Populationsgrößen kann man auch für andere Abweichungen wie überlappende Generationen finden. In der Regel ist dabei die effektiven Populationsgröße kleiner als die aktuelle Populationsgröße.
Ausgerüstet mit diesem Handwerkszeug können wir uns nun Problemen widmen, die weniger strenge Annahmen verlangen.
Wollen wir unsere Modelle nun aber auf realistischere Systeme anwenden, brauchen wir das Konzept der effektiven Populationsgröße(Ne).
Viele Population haben beispielsweise eine ungleiche Anzahl sich fortpflanzender Männchen und Weibchen. Dies ist bei starker männlicher Konkurrenz um die Weibchen der Fall, wo sich in jeder Generation nur ein Bruchteil der Männchen fortpflanzen.
In diesem Fall ist
[3]Wobei Nm die Anzahl der sich fortpflanzenden Männchen, Nf die Anzahl der sich fortpflanzenden Weibchen ist.
Spielt man etwas mit dieser Formel, wir beim Einsetzen von Werten sehr schnell deutlich, dass wenn Nm sehr viel größer als Nf der Wert für Ne eher in der Nähe des kleinen Wertes liegt. Dies macht Sinn, da durch die wenigen sich paarenden Männchen in jeder neuen Generation eine Art Flaschenhals ensteht: Die Hälfte der autosomal weitergegebenen Allele wird durch einen kleinen Bruchteil der Individuen weitergegeben.
Ähnliches gilt aus den gleichen Gründen, wenn sich einzelne Individuen -unabhängig vom Geschlecht- sehr unterschiedliche Nachkommenzahlen haben, dann ist
[4]wobei Vk die Varianz in der Nachkommenzahl ist. Da bei gleichbleibender Populationsgröße (bisher immer noch eine Ausgangsannahme) durchschnittlich zwei Nachkommen pro Elternteil entstehen, ist die Nachkommenanzahl bei zufälliger Fortpflanzung Poisson-verteilt mit einem Mittelwert und einer Varianz von 2. Größere Varianzen lassen in obiger Formel Ne kleiner als N werden.
Doch was passiert wenn sich die Populationsgröße über die Zeit ändert? Ganz einfach
[5]d.h. Ne ist das harmonische Mittel der Populationsgrößen in n Generationen.
Ähnliche Gleichungen für die effektiven Populationsgrößen kann man auch für andere Abweichungen wie überlappende Generationen finden. In der Regel ist dabei die effektiven Populationsgröße kleiner als die aktuelle Populationsgröße.
Ausgerüstet mit diesem Handwerkszeug können wir uns nun Problemen widmen, die weniger strenge Annahmen verlangen.
Mittwoch, 26. November 2008
Die neutrale Theorie der molekularen Evolution
Meine erstes Thema wird die Ausbreitung von Mutationen innerhalb einer finiten Population sein.
Motoo Kimura entwickelte seine Theorie dazu in den 1960er bis 1980er Jahren ausgehend von Anwendungen von Diffusions Approximationen auf genetische Fragestellungen, an denen zuvor R.A.Fisher und S. Wright gearbeitet hatten. Die Herleitung der Formeln übersteigt dabei mein mathematisches Verständnis. Die Theorie (und ihre nahezu neutrale Erweiterung) ist aber eine der elegantesten in der Biologie und daher auch intuitiv verständlich.
Ich versuche deshalb nur ihre Grundzüge ohne Anspruch auf Vollständigkeit darzustellen, zu zeigen welche Annahmen benötigt werden und welche Vorhersagen dies erlaubt. Bewusst wähle ich diesen Ansatz mit einer der mathematisch komplexesten Theorien zu starten (in den folgenden Posts kann es also nur einfacher werden) und werde später bei mathematisch einfacheren Theorien mehr auf Herleitung und Entwicklung der Formeln eingehen.
Hauptsächlich interessiert mich im aktuellen Post die Fixierungswahrschheinichkeit eines Allels (Ausprägungszustand eines Gens), oder spezieller einer neuen Mutation. Fixierung bedeutet hierbei, dass in der Population ausschließlich das betreffende Allel vorkommt. Der Verlust des Alles oder dessen Fixierung stellen Extremzustände da, die sich in einer vereinfachten Darstellung untersuchen lassen.
Hartl und Clark benutzen das Beispiel einer Bouling-Bahn in der die seitlichen Rinnen Analoga dieser Extremzustände sind. Nimmt man nun an, dass die -analog zur Zeit- unendlich lange Bahn -analog zu möglichen Zufallsereignissen- nicht perfekt eben ist- wird offensichtlich, dass jedes Allel über kurz oder lang einen dieser Extremzustände erreicht.
Wichtig ist lediglich die Breite der Bahn oder ihr biologisches Analogon, die Populationsgröße.
Wir nehmen eine diploide Population mit N Individuen an, in dieser sind 2N Kopie des interessierenden Gens vorhanden und es werden 2N Gameten für die nächste Generation gewählt , die dann wieder N Zygoten bilden (=gleichbleibende Populationsgröße und zufällige Paarung). Die Fixierunswahrscheinlickeit eines Alles ist nun gegeben durch seine aktuelle Frequenz p0/Anzahl der Kopien. Im Falle einer neuen Mutation, die per Definition nur einmal vorhanden ist
 [1]
[1]
Das macht Intuitiv Sinn, da jedes Gen einen Fixierungszustand ansteuert und zum Startzeitpunkt eben 2N Alternativen gegeben sind.

{1}
Betrachtet man die aus dieser Formel resultierende Fixierungswahrscheinlichkeit für eine Mutation als Funktion der Populationsgröße wird deutlich, dass diese Wahrscheinlichkeit selbst für eine moderate Populationsgröße nicht besonders groß ist. Sie ist allerdings auch nicht 0 für große Populationen (z.B. N=1,000,000-> p= 0,0000005)
u ist die Mutationsrate mit der irgendwo im interessierenden Abschnitt des Genoms eine Mutation entsteht. Neutralität kann man nun für einen ganzen Abschnitt des Genoms, wie z.B. ein Pseudogen, annehmen oder für spezielle Mutationen, wie z.B. jene von degernerierte Basen an der dritten Stelle eines Kodons (= synonyme Mutationen).
Die Rate u, mit der die Mutationen entstehen ist nun erstaunlicherweise gleich der Rate mit der neutrale Mutationen fixiert werden K. Sie ist unabhängig von der Populationsgröße, da in großen Populationen auch mehr Mutationen entstehen.
 [2]
[2]
d.h. die kleinere Fixierungswahrscheinlichkeit und die Populationsgröße heben sich gegenseitig auf. Ein Zusammenhang von mathematisch schlichter Schönheit.
Die durchschnittliche Zeit zwischen zwei Fixierungen ist dann logischerweise 1/u.
Dieses Modell passt logischerweise nicht immer zu den beobachteten Daten und ist daher sehr hilfreich als Nullhypothese um Neutralität zu testen. Es ist allerdings falsch aus abweichenden Beobachtungen auf Nicht-Neutralität zu schließen, da auch andere Voraussetzungen wie beispielsweise die gleichbleibende Populationsgröße verletzt sein können.
Folgerungen aus der neutralen Theorie der molekularen Evolution tauchen in zukünftigen Post wieder auf. In diesen werde ich näher auf den Einfluss von Selektion und damit auf die nahezu neutrale Version der Theorie eingehen, finite Populationsgrößen näher beleuchten und Zusammenhänge von Polymorphismus und Divergenz aufzeigen.
Motoo Kimura entwickelte seine Theorie dazu in den 1960er bis 1980er Jahren ausgehend von Anwendungen von Diffusions Approximationen auf genetische Fragestellungen, an denen zuvor R.A.Fisher und S. Wright gearbeitet hatten. Die Herleitung der Formeln übersteigt dabei mein mathematisches Verständnis. Die Theorie (und ihre nahezu neutrale Erweiterung) ist aber eine der elegantesten in der Biologie und daher auch intuitiv verständlich.
Ich versuche deshalb nur ihre Grundzüge ohne Anspruch auf Vollständigkeit darzustellen, zu zeigen welche Annahmen benötigt werden und welche Vorhersagen dies erlaubt. Bewusst wähle ich diesen Ansatz mit einer der mathematisch komplexesten Theorien zu starten (in den folgenden Posts kann es also nur einfacher werden) und werde später bei mathematisch einfacheren Theorien mehr auf Herleitung und Entwicklung der Formeln eingehen.
Hauptsächlich interessiert mich im aktuellen Post die Fixierungswahrschheinichkeit eines Allels (Ausprägungszustand eines Gens), oder spezieller einer neuen Mutation. Fixierung bedeutet hierbei, dass in der Population ausschließlich das betreffende Allel vorkommt. Der Verlust des Alles oder dessen Fixierung stellen Extremzustände da, die sich in einer vereinfachten Darstellung untersuchen lassen.
Hartl und Clark benutzen das Beispiel einer Bouling-Bahn in der die seitlichen Rinnen Analoga dieser Extremzustände sind. Nimmt man nun an, dass die -analog zur Zeit- unendlich lange Bahn -analog zu möglichen Zufallsereignissen- nicht perfekt eben ist- wird offensichtlich, dass jedes Allel über kurz oder lang einen dieser Extremzustände erreicht.
Wichtig ist lediglich die Breite der Bahn oder ihr biologisches Analogon, die Populationsgröße.
Wir nehmen eine diploide Population mit N Individuen an, in dieser sind 2N Kopie des interessierenden Gens vorhanden und es werden 2N Gameten für die nächste Generation gewählt , die dann wieder N Zygoten bilden (=gleichbleibende Populationsgröße und zufällige Paarung). Die Fixierunswahrscheinlickeit eines Alles ist nun gegeben durch seine aktuelle Frequenz p0/Anzahl der Kopien. Im Falle einer neuen Mutation, die per Definition nur einmal vorhanden ist
[1]Das macht Intuitiv Sinn, da jedes Gen einen Fixierungszustand ansteuert und zum Startzeitpunkt eben 2N Alternativen gegeben sind.
{1}
Betrachtet man die aus dieser Formel resultierende Fixierungswahrscheinlichkeit für eine Mutation als Funktion der Populationsgröße wird deutlich, dass diese Wahrscheinlichkeit selbst für eine moderate Populationsgröße nicht besonders groß ist. Sie ist allerdings auch nicht 0 für große Populationen (z.B. N=1,000,000-> p= 0,0000005)
u ist die Mutationsrate mit der irgendwo im interessierenden Abschnitt des Genoms eine Mutation entsteht. Neutralität kann man nun für einen ganzen Abschnitt des Genoms, wie z.B. ein Pseudogen, annehmen oder für spezielle Mutationen, wie z.B. jene von degernerierte Basen an der dritten Stelle eines Kodons (= synonyme Mutationen).
Die Rate u, mit der die Mutationen entstehen ist nun erstaunlicherweise gleich der Rate mit der neutrale Mutationen fixiert werden K. Sie ist unabhängig von der Populationsgröße, da in großen Populationen auch mehr Mutationen entstehen.
[2]d.h. die kleinere Fixierungswahrscheinlichkeit und die Populationsgröße heben sich gegenseitig auf. Ein Zusammenhang von mathematisch schlichter Schönheit.
Die durchschnittliche Zeit zwischen zwei Fixierungen ist dann logischerweise 1/u.
Dieses Modell passt logischerweise nicht immer zu den beobachteten Daten und ist daher sehr hilfreich als Nullhypothese um Neutralität zu testen. Es ist allerdings falsch aus abweichenden Beobachtungen auf Nicht-Neutralität zu schließen, da auch andere Voraussetzungen wie beispielsweise die gleichbleibende Populationsgröße verletzt sein können.
Folgerungen aus der neutralen Theorie der molekularen Evolution tauchen in zukünftigen Post wieder auf. In diesen werde ich näher auf den Einfluss von Selektion und damit auf die nahezu neutrale Version der Theorie eingehen, finite Populationsgrößen näher beleuchten und Zusammenhänge von Polymorphismus und Divergenz aufzeigen.
Dienstag, 25. November 2008
Populationsgenetik, Now!
Meine Serie
Dies ist ein Beitrag in einer Reihe von Posts zu Populations- und quantitativer Genetik. Es gibt im deutschsprachigen Raum meines Wissens kein Lehrbuch zu diesem Thema. Dies ist wohl eine der Folgen des zu niedrigen Stellenwertes der Evolutionsbiologie an deutschen Hochschulen, wie ihn auch der VBIO beklagt.
Eine anderer möglicher Grund für die fehlende "quantitative Tradition" in der deutschen Evolutionsbiologie ist vielleicht auch, dass der bekannteste deutschsprachige Vertreter dieser Disziplin, Ernst Mayr nicht mit mathematischen Modellen arbeitete.
Die Posts dieser Reihe werde ich hauptsächlich mit Hilfe der Bücher "Principles of population genetics" von Daniel L Hartl und Andrew G. Clark, "Quantitative genetics" von Douglas S. Falconer und Trudy F.C. Mackay schreiben. Außerdem habe ich in den letzten Monaten eine Vorlesung bei Brian Charlsworth und Peter Keightley besucht, die Skripte und Aufzeichnungen aus diesen werde ich ebenfalls konsultieren.
Trotzdem werden die Posts natürlich nur einen winzigen Einblick in das große Feld verschaffen und sicher auch Fehler enthalten.
Warum sollte man sich gerade jetzt mit Populationgenetik beschäftigen?
In einem interessanten Post auf dem Fischblog beschreibt Godwael den großen zu erwartenden Erkenntnisgewinn aus der Sequenzierung hunderter kompletter menschlicher Genome. Dabei ist mir aufgefallen, dass die theoretischen Grundlagen der Populationsgenetik im deutschsprachigen Raum wohl eher unbekannt sind.
Wie breiten sich Mutationen aus? Wie ausgeprägt sind die Einflüsse von Migration, Drift und Selektion? All diese Fragestellungen müssen nicht anhand der an kompletten Genomsequenzen gewonnenen Daten untersucht werden, sondern es existiert eine unglaubliche Fülle an Modellen, die das das Zusammenspiel dieser Faktoren testen. Natürlich ist nicht auszuschließen, dass auch neue Modele entwickelt werden müssen, das Gros der neu gewonnenen Daten passt aber zu den bestehenden Erklärungsansätzen.
Welchen Nutzen ziehen Evolutionsbiologen also aus den neu gewonnenen Genom-Daten?
Einer der Hauptnutzen besteht darin, dass sie die Suche nach den am besten passenden Modellen für bislang ununtersuchte Genombereiche erlauben. Evolviert ein Bereich des Genoms dann anders als man es unter einem bestimmten Modell erwarten würde, ist die Verwendung eines anderen Modells mit veränderten Ausgangs-Annahmen nötig. Hat man dann ein Modell gefunden das die Daten anhand der der sparsamsten Parameter (Occam's Razor) bestmöglich beschreibt generiert dies wiederum neue Hypothesen.
Beispielsweise könnte es notwendig werden über historisch noch unbekannte Migrationsbewegungen menschlicher Populationen nachzudenken oder Selektion auf einen Bereich des Genoms in Betracht zu ziehen der zuvor als neutral galt. Je nachdem was die Modelle nahelegen können so beispielsweise Hypothesen für Historiker, Zellbiologen oder Biochemiker generiert werden. Die Fähigkeit der entsprechenden Wissenschaftler diese Implikationen der Evolutionsbiologie für ihr Forschungsfeld zu verstehen wird in einigen Beriechen sicher Entdeckungen fördern. Es ist also für viele Wissenschaftler ratsam sich in nächster Zeit etwas mit theoretischer Evolutionsbiologie zu beschäftigen.
Dies ist ein Beitrag in einer Reihe von Posts zu Populations- und quantitativer Genetik. Es gibt im deutschsprachigen Raum meines Wissens kein Lehrbuch zu diesem Thema. Dies ist wohl eine der Folgen des zu niedrigen Stellenwertes der Evolutionsbiologie an deutschen Hochschulen, wie ihn auch der VBIO beklagt.
Eine anderer möglicher Grund für die fehlende "quantitative Tradition" in der deutschen Evolutionsbiologie ist vielleicht auch, dass der bekannteste deutschsprachige Vertreter dieser Disziplin, Ernst Mayr nicht mit mathematischen Modellen arbeitete.
Die Posts dieser Reihe werde ich hauptsächlich mit Hilfe der Bücher "Principles of population genetics" von Daniel L Hartl und Andrew G. Clark, "Quantitative genetics" von Douglas S. Falconer und Trudy F.C. Mackay schreiben. Außerdem habe ich in den letzten Monaten eine Vorlesung bei Brian Charlsworth und Peter Keightley besucht, die Skripte und Aufzeichnungen aus diesen werde ich ebenfalls konsultieren.
Trotzdem werden die Posts natürlich nur einen winzigen Einblick in das große Feld verschaffen und sicher auch Fehler enthalten.
Warum sollte man sich gerade jetzt mit Populationgenetik beschäftigen?
In einem interessanten Post auf dem Fischblog beschreibt Godwael den großen zu erwartenden Erkenntnisgewinn aus der Sequenzierung hunderter kompletter menschlicher Genome. Dabei ist mir aufgefallen, dass die theoretischen Grundlagen der Populationsgenetik im deutschsprachigen Raum wohl eher unbekannt sind.
Wie breiten sich Mutationen aus? Wie ausgeprägt sind die Einflüsse von Migration, Drift und Selektion? All diese Fragestellungen müssen nicht anhand der an kompletten Genomsequenzen gewonnenen Daten untersucht werden, sondern es existiert eine unglaubliche Fülle an Modellen, die das das Zusammenspiel dieser Faktoren testen. Natürlich ist nicht auszuschließen, dass auch neue Modele entwickelt werden müssen, das Gros der neu gewonnenen Daten passt aber zu den bestehenden Erklärungsansätzen.
Welchen Nutzen ziehen Evolutionsbiologen also aus den neu gewonnenen Genom-Daten?
Einer der Hauptnutzen besteht darin, dass sie die Suche nach den am besten passenden Modellen für bislang ununtersuchte Genombereiche erlauben. Evolviert ein Bereich des Genoms dann anders als man es unter einem bestimmten Modell erwarten würde, ist die Verwendung eines anderen Modells mit veränderten Ausgangs-Annahmen nötig. Hat man dann ein Modell gefunden das die Daten anhand der der sparsamsten Parameter (Occam's Razor) bestmöglich beschreibt generiert dies wiederum neue Hypothesen.
Beispielsweise könnte es notwendig werden über historisch noch unbekannte Migrationsbewegungen menschlicher Populationen nachzudenken oder Selektion auf einen Bereich des Genoms in Betracht zu ziehen der zuvor als neutral galt. Je nachdem was die Modelle nahelegen können so beispielsweise Hypothesen für Historiker, Zellbiologen oder Biochemiker generiert werden. Die Fähigkeit der entsprechenden Wissenschaftler diese Implikationen der Evolutionsbiologie für ihr Forschungsfeld zu verstehen wird in einigen Beriechen sicher Entdeckungen fördern. Es ist also für viele Wissenschaftler ratsam sich in nächster Zeit etwas mit theoretischer Evolutionsbiologie zu beschäftigen.
Seeigelsex Protokoll
Als Ergaenzung zu Argent23's Post "Seeigelsex" auf Holiday Junction poste ich hier das original Protokoll der Giglio-Exkursion von 2006. Es soll ja Leute geben, die ein Mikroskop ihr eigen nennen und unter dieser einen Vorraussetzungen ist das Ganze eine wirklich spannende Urlaubsbeschaeftigung. Vielleicht stolpert ja auch der eine oder andere Kursteilnehmer bei der Vorbereitung der Versuche ueber diese Posts... Viel Spass!
Larvalentwicklung der Echinodermata
1. Einleitung
Stachelhäuter sind getrenntgeschlechtlich und pflanzen sich fort, indem sie Samenzellen und Eier direkt ins Wasser freisetzen. Das Geschlechterverhältnis ist dadurch bei vielen Arten zugunsten der Männchen auf etwa Drei zu Eins verschoben. Die meisten Arten haben pelagische Larven, die sich von Plankton ernähren. Im Gegensatz zu ihren Eltern sind die Larven bilateralsymmetrisch. Erst wenn sie sich auf dem Boden niederlassen verändert sich ihr Körper und zeigt die typische Radiärsymmetrie. In der Entwicklung der Larven wird außerdem der Urmund zum späteren After, und ein sekundärer Mund bildet sich, was die Echinodermata als Deuterostomier ausweist.
Seeigelembryonen werden daher, und weil sie leicht zugänglich und durchsichtig sind als Modellsystem für Entwicklungsvorgänge benutzt.
2.Gewinnung der Gameten
Wir benutzten für unseren Versuch den Schwarzen Seeigel, Arbacia lixula, der im Mittelmeerraum und so auch auf Giglio an allen Felsküsten häufig vorkommt.
Da die Tiere sehr empfindlich auf Kontakt mit der Luft reagieren, wurde einmal sofort am Strand versucht Eizellen und Sperma zu gewinnen, ein zweites Mal wurden andere Tiere erst im Labor der Prozedur unterzogen. Die Tiere wurden mit der Oralseite nach oben in ein Becherglas, gefüllt mit sterilfiltriertem Meerwasser, gesetzt. Zum Sterilfiltrieren hatten wir das Meerwasser mit einer Spritze und einem geeigneten Aufsatz durch einen Filter mit einer Maschenweite von 0,2μm gepresst.
Den Seeigeln wurde nun etwa 2ml Kaliumchlorid mit einer Injektionsspritze durch die Mundöffnung in die Leibeshöhle injiziert, was eine Depolarisation und ein Ausstoßen der Gameten bewirken sollte.
Dazu verwendeten wir am Strand versehentlich 30%iges (circa 5M) im Labor später 0,5 molares KCl. Trotzdem kam es bei beiden Ansätzen zu einem Ausstoßen von Gameten, wobei nicht alle Tiere wie gewünscht reagierten. Es zeigte sich dass eine schräge Injektion tief ins innere des Tieres die größte
Erfolgswahrscheinlichkeit mit sich bringt. Dies könnte daher rühren, dass so das Kaliumchlorid direkt in die Gonade seitlich unter der Apikalseite gespritzt wurde.

Folgende Tabellen zeigen die Anzahl männlicher und weiblicher Tiere, die Keimzellen ausstießen. Die Zahl in der Spalte BG gibt an wie viele Tiere weder Spermien noch Eizellen abgaben.
Strand
BG---männlich---weiblich
6------------3---------------2
Labor
BG---männlich---weiblich
4------------4---------------2
Durch die hohe Anzahl von Tieren unbestimmten Geschlechts macht eine Statistik der Geschlechterverteilung keinen Sinn. Die Gameten wurden mit einer Pipette vom Boden der Bechergläser entnommen und getrennt, gekühlt aufbewahrt.
Entwicklung der Embryonen
Wir gaben nun in zwei Petrischalen die am Strand erhaltenen Spermien zu den dort gewonnenen Eizellen und stellten eine der beiden Schalen kühl.
Ebenso verfuhren wir mit den erst im Labor erhaltenen Geschlechtszellen.
Nach der Befruchtung der bereits polar gebauten Eizelle(Polkörperchen am animalen Pol) wurde ein Eindringen weiterer Spermien durch eine Veränderung der Plasmamembran zur Befruchtungsmembran, die nun so genannte Corticalgranula als äußere Schutzschicht enthält, verhindert. Diese Befruchtungsmembran war bei allen Versuchsansätzen als dünner Kranz um die Zygote zu erkennen.

Auch die ersten beiden Furchungen, die das Ei entlang der animal-vegetativen Achse teilen konnten wir bei den bei Zimmertemperatur gelagerten Ansätzen nach ca.20 und 40 Minuten erkennen. Die im Kühlschrank gelagerten Embryonen erreichten nach etwa 20 Stunden dieses Stadium.

Nach etwa zwei Stunden hatten die Zimmertemperatur-Ansätze das 16-Zell-Stadium erreicht. Dieses entsteht nach der vierten inäqual verlaufenden Furchung und zeigt am vegetativen Pol vier kleine Mikromeren.

Nach etwa 5 Stunden befanden sich in den beiden bei Zimmertemperatur gelagerten Ansätzen bereits Blastulae. Auch die gekühlten Ansätze erreichten dieses Stadium, allerdings erst nach fünf Tagen.

Die darauf folgenden Stadien, zwischen Blastula und Larve, entwickelten sich über Nacht und konnten daher leider nicht dokumentiert werden.

Bei Beginn der Gastrulation wanderten vom vegetativen Pol etwa 40 primäre
Mesenchymzellen in das innere der Blastula. Dort stülpte sich der Darm ein, der dann mit dem Mund, der sich von der gegenüberliegenden Seite eingestülpt hatte, verschmolz.
Nach etwa 48 Stunden konnten wir im Ansatz, mit den am Strand gewonnenen
Gameten, der bei Zimmertemperatur gelagert wurde, Pluteuslarven entdecken.

Die Embryonen des im Labor gewonnenen Ansatzes waren von Pilzsporen oder
anderen Verunreinigungen zerstört. Auch die beiden im Kühlschrank gelagerten Ansätze starben nach fünf Tagen im Blastulastadium ab.
Wahrscheinlich war nur beim ersten Sterilfiltrieren des Meerwassers unser Filteraufsatz richtig dicht.
In unserem ersten Ansatz konnten wir nach drei Tagen sogar die beginnende
Metamorphose zum radiärsymmetrischen Tier beobachten. Es hatte sich am Boden der Petrischale mit einem radiärsymmetrischen Auswuchs angeheftet.

Larvalentwicklung der Echinodermata
1. Einleitung
Stachelhäuter sind getrenntgeschlechtlich und pflanzen sich fort, indem sie Samenzellen und Eier direkt ins Wasser freisetzen. Das Geschlechterverhältnis ist dadurch bei vielen Arten zugunsten der Männchen auf etwa Drei zu Eins verschoben. Die meisten Arten haben pelagische Larven, die sich von Plankton ernähren. Im Gegensatz zu ihren Eltern sind die Larven bilateralsymmetrisch. Erst wenn sie sich auf dem Boden niederlassen verändert sich ihr Körper und zeigt die typische Radiärsymmetrie. In der Entwicklung der Larven wird außerdem der Urmund zum späteren After, und ein sekundärer Mund bildet sich, was die Echinodermata als Deuterostomier ausweist.
Seeigelembryonen werden daher, und weil sie leicht zugänglich und durchsichtig sind als Modellsystem für Entwicklungsvorgänge benutzt.
2.Gewinnung der Gameten
Wir benutzten für unseren Versuch den Schwarzen Seeigel, Arbacia lixula, der im Mittelmeerraum und so auch auf Giglio an allen Felsküsten häufig vorkommt.
Da die Tiere sehr empfindlich auf Kontakt mit der Luft reagieren, wurde einmal sofort am Strand versucht Eizellen und Sperma zu gewinnen, ein zweites Mal wurden andere Tiere erst im Labor der Prozedur unterzogen. Die Tiere wurden mit der Oralseite nach oben in ein Becherglas, gefüllt mit sterilfiltriertem Meerwasser, gesetzt. Zum Sterilfiltrieren hatten wir das Meerwasser mit einer Spritze und einem geeigneten Aufsatz durch einen Filter mit einer Maschenweite von 0,2μm gepresst.
Den Seeigeln wurde nun etwa 2ml Kaliumchlorid mit einer Injektionsspritze durch die Mundöffnung in die Leibeshöhle injiziert, was eine Depolarisation und ein Ausstoßen der Gameten bewirken sollte.
Dazu verwendeten wir am Strand versehentlich 30%iges (circa 5M) im Labor später 0,5 molares KCl. Trotzdem kam es bei beiden Ansätzen zu einem Ausstoßen von Gameten, wobei nicht alle Tiere wie gewünscht reagierten. Es zeigte sich dass eine schräge Injektion tief ins innere des Tieres die größte
Erfolgswahrscheinlichkeit mit sich bringt. Dies könnte daher rühren, dass so das Kaliumchlorid direkt in die Gonade seitlich unter der Apikalseite gespritzt wurde.
Folgende Tabellen zeigen die Anzahl männlicher und weiblicher Tiere, die Keimzellen ausstießen. Die Zahl in der Spalte BG gibt an wie viele Tiere weder Spermien noch Eizellen abgaben.
Strand
BG---männlich---weiblich
6------------3---------------2
Labor
BG---männlich---weiblich
4------------4---------------2
Durch die hohe Anzahl von Tieren unbestimmten Geschlechts macht eine Statistik der Geschlechterverteilung keinen Sinn. Die Gameten wurden mit einer Pipette vom Boden der Bechergläser entnommen und getrennt, gekühlt aufbewahrt.
Entwicklung der Embryonen
Wir gaben nun in zwei Petrischalen die am Strand erhaltenen Spermien zu den dort gewonnenen Eizellen und stellten eine der beiden Schalen kühl.
Ebenso verfuhren wir mit den erst im Labor erhaltenen Geschlechtszellen.
Nach der Befruchtung der bereits polar gebauten Eizelle(Polkörperchen am animalen Pol) wurde ein Eindringen weiterer Spermien durch eine Veränderung der Plasmamembran zur Befruchtungsmembran, die nun so genannte Corticalgranula als äußere Schutzschicht enthält, verhindert. Diese Befruchtungsmembran war bei allen Versuchsansätzen als dünner Kranz um die Zygote zu erkennen.
Auch die ersten beiden Furchungen, die das Ei entlang der animal-vegetativen Achse teilen konnten wir bei den bei Zimmertemperatur gelagerten Ansätzen nach ca.20 und 40 Minuten erkennen. Die im Kühlschrank gelagerten Embryonen erreichten nach etwa 20 Stunden dieses Stadium.
Nach etwa zwei Stunden hatten die Zimmertemperatur-Ansätze das 16-Zell-Stadium erreicht. Dieses entsteht nach der vierten inäqual verlaufenden Furchung und zeigt am vegetativen Pol vier kleine Mikromeren.
Nach etwa 5 Stunden befanden sich in den beiden bei Zimmertemperatur gelagerten Ansätzen bereits Blastulae. Auch die gekühlten Ansätze erreichten dieses Stadium, allerdings erst nach fünf Tagen.
Die darauf folgenden Stadien, zwischen Blastula und Larve, entwickelten sich über Nacht und konnten daher leider nicht dokumentiert werden.
Bei Beginn der Gastrulation wanderten vom vegetativen Pol etwa 40 primäre
Mesenchymzellen in das innere der Blastula. Dort stülpte sich der Darm ein, der dann mit dem Mund, der sich von der gegenüberliegenden Seite eingestülpt hatte, verschmolz.
Nach etwa 48 Stunden konnten wir im Ansatz, mit den am Strand gewonnenen
Gameten, der bei Zimmertemperatur gelagert wurde, Pluteuslarven entdecken.
Die Embryonen des im Labor gewonnenen Ansatzes waren von Pilzsporen oder
anderen Verunreinigungen zerstört. Auch die beiden im Kühlschrank gelagerten Ansätze starben nach fünf Tagen im Blastulastadium ab.
Wahrscheinlich war nur beim ersten Sterilfiltrieren des Meerwassers unser Filteraufsatz richtig dicht.
In unserem ersten Ansatz konnten wir nach drei Tagen sogar die beginnende
Metamorphose zum radiärsymmetrischen Tier beobachten. Es hatte sich am Boden der Petrischale mit einem radiärsymmetrischen Auswuchs angeheftet.
Donnerstag, 20. November 2008
Statistik ist mehr als die drei Tests, und was ich im Moment so mache.
Der Hauptgrund warum hier nichts los ist hat nur einen Buchstaben: R.
Während meiner Diplomarbeit habe ich neben meinem Interesse an Genomik auch einen Hang zu quantitativem Arbeiten entwickelt. Genauer gesagt sollte ich damals die Darmwand "meines [1]" Wurms elektronenmikroskopisch untersuchen und verschiedene Parameter dieses Epithels aus verschiedenen "experimentellen Gruppen" vergleichen(d vermessen). Ich habe mir daraufhin ein Programm beschafft mit dem ich die digitalisierten Bilder vermessen konnte und habe so eine riesigen Datensatz generiert.
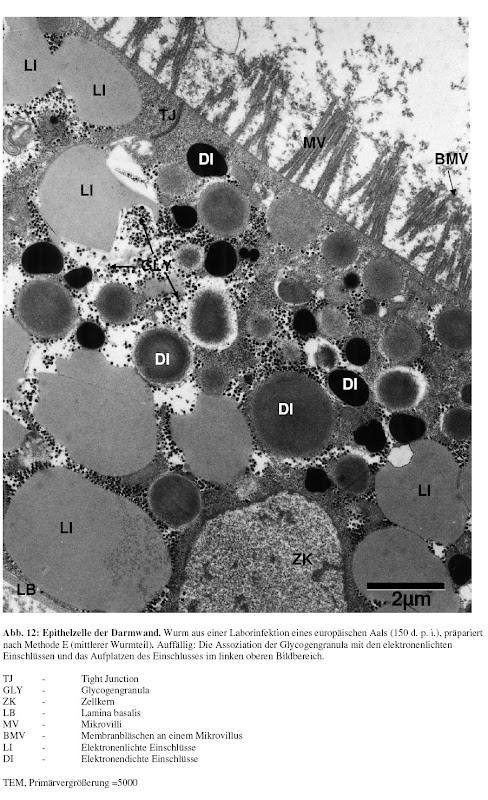
Die Analyse in SPSS gestaltete sich dann folgendermaßen:
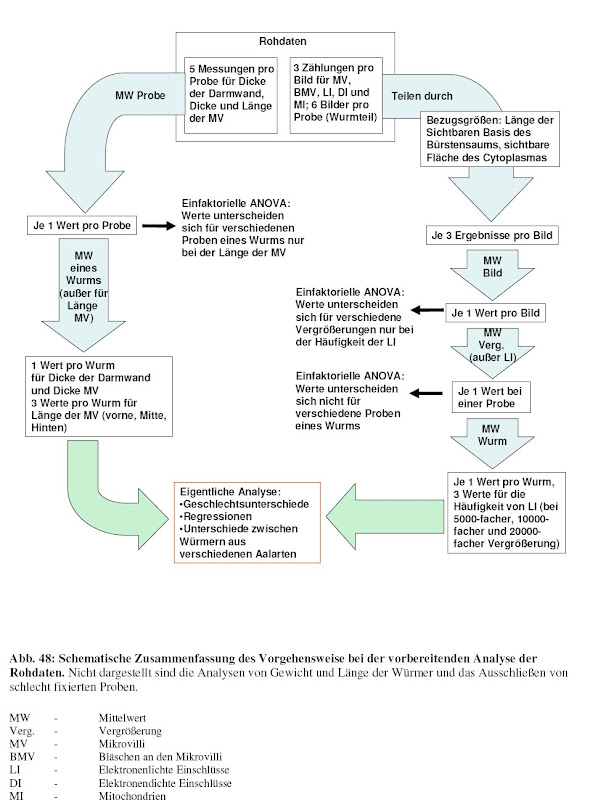
Alles was nach den erste beiden Pfeilen steht ist dabei was die Vorgehensweise bei der Datenauswertung betrifft bestenfalls nicht besonders elegant [2]. Effekte aller Variablen habe ich nacheinander in willkürlicher Weise auseinander "seziert".
Die korrekte Vorgehensweise wäre ein statistisches Modell auf die Daten anzupassen. Dabei würden dann schrittweise von einem Maximalmodell ausgehend nicht signifikante Variablen ausgeschlossen.
Einer der vielen Vorteile von R ist nun, dass es die Datenanalyse geradezu in Richtung einer solchen Modellierung lenkt.
Der Datensatz meiner Diplomarbeit ist für meine derzeitigen Fähigkeiten trotzdem noch etwas zu kompliziert. Ich beschäftige mich hauptsächlich mit Infektionsdaten, die ich Anfang des Jahres in Taiwan gesammelt habe. Es geht darum den Zusammenhang von Kapseln in der Darm- und Schwimmblasenwand mit der Infektion zu testen (die Kapseln sind tote Larven!).

Neben meiner statistischen Modellierung amplifizieren und sequenzieren wir ("mein" Diplomand Dominik) im Moment aus den Kapseln einige Gene für die wir sehr Nematoden-spezifische Primer haben.
[1] Sobald "mein" Transkriptom online ist lass ich die Anführungszeichen weg ;-)
[2] Ich wurde gerade dafür trotzdem von den Gutachtern meiner Arbeit gelobt. Das Hauptproblem an diesem Datensatz ist auch wirklich die pseudo-Replikation, darum die vielen Mittelwerte.
Während meiner Diplomarbeit habe ich neben meinem Interesse an Genomik auch einen Hang zu quantitativem Arbeiten entwickelt. Genauer gesagt sollte ich damals die Darmwand "meines [1]" Wurms elektronenmikroskopisch untersuchen und verschiedene Parameter dieses Epithels aus verschiedenen "experimentellen Gruppen" vergleichen(d vermessen). Ich habe mir daraufhin ein Programm beschafft mit dem ich die digitalisierten Bilder vermessen konnte und habe so eine riesigen Datensatz generiert.
Die Analyse in SPSS gestaltete sich dann folgendermaßen:
Alles was nach den erste beiden Pfeilen steht ist dabei was die Vorgehensweise bei der Datenauswertung betrifft bestenfalls nicht besonders elegant [2]. Effekte aller Variablen habe ich nacheinander in willkürlicher Weise auseinander "seziert".
Die korrekte Vorgehensweise wäre ein statistisches Modell auf die Daten anzupassen. Dabei würden dann schrittweise von einem Maximalmodell ausgehend nicht signifikante Variablen ausgeschlossen.
Einer der vielen Vorteile von R ist nun, dass es die Datenanalyse geradezu in Richtung einer solchen Modellierung lenkt.
Der Datensatz meiner Diplomarbeit ist für meine derzeitigen Fähigkeiten trotzdem noch etwas zu kompliziert. Ich beschäftige mich hauptsächlich mit Infektionsdaten, die ich Anfang des Jahres in Taiwan gesammelt habe. Es geht darum den Zusammenhang von Kapseln in der Darm- und Schwimmblasenwand mit der Infektion zu testen (die Kapseln sind tote Larven!).
Neben meiner statistischen Modellierung amplifizieren und sequenzieren wir ("mein" Diplomand Dominik) im Moment aus den Kapseln einige Gene für die wir sehr Nematoden-spezifische Primer haben.
[1] Sobald "mein" Transkriptom online ist lass ich die Anführungszeichen weg ;-)
[2] Ich wurde gerade dafür trotzdem von den Gutachtern meiner Arbeit gelobt. Das Hauptproblem an diesem Datensatz ist auch wirklich die pseudo-Replikation, darum die vielen Mittelwerte.
Samstag, 8. November 2008
Das Volk hat gesprochen!
Ich beuge mich einer eindeutigen 2/3 Mehrheit und werde weiterhin auf Deutsch, Englisch und Denglisch bloggen! Sobald ich wieder dazu komme...
Sonntag, 2. November 2008
Best nematode quote ever
NATHAN AUGUSTUS COBB
1859-1932
"In short, if all the matter in the universe except the
nematodes were swept away, our world would still be dimly
recognizable, and if, as disembodied spirits, we could then
investigate it, we should find its mountains, hills, vales,
rivers, lakes, and oceans represented by a film of
nematodes. The location of towns would be decipherable,
since for every massing of human beings there would be a
corresponding massing of certain nematodes. Trees would
still stand in ghostly rows representing our streets and
highways. The location of the various plants and animals
would still be decipherable, and, had we sufficient
knowledge, in many cases even their species could be
determined by an examination of their erstwhile nematode
parasites."
from "Nematodes and Their Relationships", 1915
Mittwoch, 29. Oktober 2008
VW symposium: Abstract
My abstract for the VW Symposium:
Anguillicola crassus is an eel-specific swimbladder nematode, which parasitizes the Japanese eel Anguilla japonica, found throughout East Asia. This species has also colonized a number of novel hosts: since the early 1980s it has spread throughout almost all populations of the European eel Anguilla anguilla, and also the American eel A. rostrata, since the 1990s. Morphological divergence of the European vs. Asian populations of A. crassus has been documented in field studies. Cross infection experiments, comparing the degree of divergence between the two nematode populations harbored in the same host species, suggest a generally decreased virulence of the European population in comparison to the Asian population, as well as differentiation of live history traits. We are currently employing new high throughput sequencing technology and analysis of gene expression data to identify the potential underlying genes for these differences, with the aim to elucidate whether divergence is driven by evolution of gene expression or coding sequence. We are also testing the "adaptationist" hypothesis that the identified genes may be involved in host-parasite interaction. Finally we will test whether these candidate genes also play a role in divergence in introduced populations of A. crassus in American eels.
Anguillicola crassus is an eel-specific swimbladder nematode, which parasitizes the Japanese eel Anguilla japonica, found throughout East Asia. This species has also colonized a number of novel hosts: since the early 1980s it has spread throughout almost all populations of the European eel Anguilla anguilla, and also the American eel A. rostrata, since the 1990s. Morphological divergence of the European vs. Asian populations of A. crassus has been documented in field studies. Cross infection experiments, comparing the degree of divergence between the two nematode populations harbored in the same host species, suggest a generally decreased virulence of the European population in comparison to the Asian population, as well as differentiation of live history traits. We are currently employing new high throughput sequencing technology and analysis of gene expression data to identify the potential underlying genes for these differences, with the aim to elucidate whether divergence is driven by evolution of gene expression or coding sequence. We are also testing the "adaptationist" hypothesis that the identified genes may be involved in host-parasite interaction. Finally we will test whether these candidate genes also play a role in divergence in introduced populations of A. crassus in American eels.
Promoting Science-Blogging!
Ich werde Ende Februar am "VW Foundation Evolutionary Biology Status Symposium" in Muenster teilnehmen.
Es gibt die Moeglichkeit Diskussionsgruppen (auch ausserhalb streng wissenschaftlicher Themenbereiche) vorzuschlagen, als habe ich eben an die Organisatoren geschrieben:
Mal gespannt wie der Vorschlag ankommt. Das ganze boete natuerlich die Moeglichkeit zu schamloser Eigenwerbung...
Es gibt die Moeglichkeit Diskussionsgruppen (auch ausserhalb streng wissenschaftlicher Themenbereiche) vorzuschlagen, als habe ich eben an die Organisatoren geschrieben:
I have registered for the symposium a few days ago. I just had an idea for a discussion group.
"Science 2.0: Blogging as a new way of science communication"All the best,
- A blog as a medium to publish your thoughts and for scientific discussion
- Examples of the English speaking science-blogging community
- Future directions, interaction with classical ways of science communication and publishing
Emanuel
Mal gespannt wie der Vorschlag ankommt. Das ganze boete natuerlich die Moeglichkeit zu schamloser Eigenwerbung...
Mittwoch, 22. Oktober 2008
The Brian Charlsworth quote of the week
"That`s the magic of population genetics: always take 4 times the effective population size times something!"
Sonntag, 12. Oktober 2008
The Brian Charlsworth quotes of the week
Multiple quotes, all from Thursday.
"It is like in Monty Python`s "The life of Brian": What have mutations ever done for you?"Upright walking? Increased brain size? :-)
"Fortunately the most of my genome is not doing anything...
...it`s just there for decoration!?"
...bringing a ln in the equation for the survival probability of a favourable mutation.
"So we saved the theory of natural selection from death by initial frequencies!"
We are all half dead!
...I will try to make the discussion both accessible for the interested reader and deep enough to meet the criteria of a good essay for the course.
Back to the topic. What do I mean with the title of this post?
With "we" I mean all animals and with "half dead" the fact that the average animal carries (heterozygote) more than one recessive allele that would be lethal if homozygote. The lethal allele has no influence on fitness of heterozygotes (half dead means fully alive here;-)) and reduces homozyygote fintness to 0.
You don`t need fancy technology to figure this out and the methods used for the study of McCune et al. are nearly as old as the field of quantitative genetics itself (the reference describing the method is in fact from 1927 and not accessible online). The experimentator mates simply siblings resulting from a cross of wild-caught animals and records the zygotes or embryos with developmental distortions leading to death. Sofar this seems facile, the only difficulty in interpretation of the reults is easy to resolve: If similar phenotyps are observed in different crosses the phenotypically healthy siblings from both crosses are outbred with each other. When all the ofspring of this controll is healthy two different recessive lethal allels were found.
This method has a single severe downside: In animals that have a reduced rate of survival as embryos or zygotes due to chance or environmental influences the experiments are not possibele. Therefore Xenopus laevis was the only vertebrate for which data on R (the number of recessive lethals per individual) was investigated by this method before. Extensive data is in contrast available on R for Drosophila.
McCune et al. found, that in both teleost fish species (Lucania goodei and Danio rerio) R is of comparable size to R in Drosophila.
The title "A Low Genomic Number of Recessive Lethals in Natural Populations of Bleufin Killifish and Zebrafish" is already part of their interpretation. They suggest that vertebrates have a higher number of genes and therefore R is smaller in relation to the number of sites that can cause lethal phenotypes.
McCune et al. infer the number of genes in their fishes from the number of genes in humans. They postulate that, because of the high synteny between vertebrates this number would be approximately the same. Unfortunately the estimation for the number of human genes was 35,000 back in 2002, the real number based on newer estimates is not higher than 25,000. The ratio of Drosophila/vertebrate genes comes down from 2.5 to 1.79 considering this.
McCune et al. propose the smaller size of vertebrate populations as a reason for the lower R (in relation to the number of genes) in vertebrates compared to invertebrates. This sounds intuitively right, because smaller populations result in higher inbreeding. For this reason selection against recessive lethal alleles would be more effective in the smaller vertebrate populations.
Nevertheless I have other doubts regarding the plausibility of the assumptions that R must be set in relation to the "exome-size". There would be no need for correcting with the number of genes, if all animals had a set of genes comparable in size, essential for their development. As R is the same in all animals the whole discussion (and the title) of the paper would make no sense in this context.
P.S. I am not aware whether vertebrates and invertebrates have this comparably large set of essential genes. It seems not to be known yet...
...or I should search harder.
Amy R. McCune, Rebecca C. Fuller, Allisan A. Aquilina, Robert M. Dawley, James M. Fadool, David Houle, Joseph Travis, and Alexey S. Kondrashov (2002) A Low Genomic Number of Recessive Lethals in Natural Populations of Bluefin Killifish and Zebrafish. Science 296 (5577), 2398.
[DOI: 10.1126/science.1071757]
Dienstag, 7. Oktober 2008
The Brian Charlsworth quote of the week
Ich bin sehr froh hier in Edinburgh Vorlesungen eines der bedeutendsten lebenden Evolutionsbiologen hören zu können. Und da der gute Mann immer für einen Lacher gut ist möchte ich hier jede Woche ein Zitat präsentieren. Für letzte Woche:
Die Woche davor war der beste Satz:
"Protein-biochemists don't understand proteins"Gemeint war, dass Proteinchemiker nicht verstehen, wie (dass) Selektionsdrücke auch auf scheinbar "gleichartige" (z.B. basische AS-> basische AS) Änderungen der Aminosäuresequenz wirken können. Das Zitat gefällt mir besonders, da man an meiner Heimatuni Karlsruhe fast nur von solchen umgeben ist.
Die Woche davor war der beste Satz:
"Zoologists are generally even more stupid than geneticists"Dieser Ausspuch gehörte in den Zusammenhang, dass viele Zoologen bis Mitte des letzten Jahrhunderts noch an der Vereerbung erworbener Merkmale und an "blending inheritance" festhielten.
Samstag, 27. September 2008
The Tuesday Nematode
Mit Gnathostoma spinigerum präsentiere ich heute zum ersten Mal in der "weekly Nematode"-Serie (ich sollte die Serie vielleicht mit etwas understatement in "The annual Nematode" umbenennen) einen spirurinen Nematoden. Spiruina bildet eine Schwestergruppe der Tylenchina (die beiden letzen Nematoden der Woche) und Rhabditina (meist freilebend; z.B. C. elegans). Diese Klade (Spirurina wird häufig auch einfach "Clade 3" genannt) wude erstmals 1998 entdeckt und umfasst die beiden früher auf Ordnungs-Ebene geführten Kladen Spirurida in Ascaridida.
Besonders interessant ist die Gattung Gnathostoma tatsächlich durch ihre phylogenetische Position. Da ich zurzeizeit Hilfe von einem Diplomanden habe, der sich um die Phylogeny der Gattung Anguillicola und um eine bessere Auflösung des Baums für die basalen Spirurina kümmern soll, gibt es hier eine Neuheit- einen Gastpost "meines" Diplomanden Dominik Laetsch:
Grund für unser Interesse an der Gattung Gnathostoma ist also -wie gesagt- zum einen die geringe phylogenetische Distanz der SSU-rDNA-Sequenz dieses Nematoden zu jener unseres "Lieblings-Wurms" Anguillicola crassus und die Tatsache, dass die Familien Gnathostomatidae und Aguillicolidae aufgrund jener Daten den basalen Zweig der Spirurina darzustellen scheinen [1], [2]. Bestimmte Vertreter der Gattung Gnathostoma werden uns daher auch als "Out-group" in der phylogenetischen Analyse des Genus Anguillicola dienen (was das Thema meiner Diplomarbeit darstellt).
Der Genus Gnathostoma gliedert sich in ca. 9 mehr oder weniger gut von einander abgegrenzten Arten, welche wie alle Spirurina parasitisch leben. Davon sind mindestens drei als humanpathogene Arten beschrieben: G. turgidum, G. doloresi und G. spinigerum. Der Lebenszyklus von G. spinigerum ist bekannt und umfasst die Entwicklung der L1-Larve zum, für den Endwirt infektiösen, dritten Larvenstadium in zwei aquatischen Zwischenwirten (Copepoden des Genus Cyclops bzw. Fische/ Amphibia/ Mollusca) und die anschliessende Entwicklung zum Adultus im Endwirt (Mammalia; oft Feliden, Caniden und Suiden). Paratenische Wirte stellen vor allem Fische und Vögel dar. Im Endwirt durchbohrt G. spinigerum nach der oralen Aufnahme infizierten Gewebes die Magenwand und bereist als Larva migrans für die nächsten 3 Monate dessen Gewebe und innere Organ bis er zurückkehrt und sich an die mucosale Magenwand heftet. Dort entwickelt er sich für weitere 6 Monate bis er beginnt unembryonierter Eier zu produzieren, welche durch mit dem Kot ausgeschieden werden. Im Wasser embryonieren diese Eier, und mit der Azfnahme durch den ersten Zwischenwirt ist der Kreislauf geschlossen.
Das klinische Bild wird unter dem Begriff Gnathostomiasis [3] zusammengefasst und kann sowohl bei Feliden, Caniden als auch Hominiden letal verlaufen. Erstmals beschrieben wurde dieses Krankheitsbild 1835 anhand eines Kadavers eines jungen Tigers des Londoner Zoos. In Menschen entwickelt sich G. spinigerum nicht zum Adultus sondern migriert durch das Körpergewebe für bis zu 10- 12 Jahre, wobei zwischen kutaner und viszeraler Larva migrans unterschieden wird. Ersteres beschreibt nur Hautexzesse, bedingt durch mechanische Zerstörung des Gewebes und Produktion von Proteasen, Hyaluronidasen und Hemolysin durch den Parasiten, sowie durch die Immunreaktion des Wirtes. Letzteres umfasst die selben Faktoren jedoch in Organen wie der Leber und dem ZNS. Diese Scäden führen zu einer Mortalität von 8-25 % der betrofenen Patienten. Desweiteren gibt es drei dokumentierte Fälle von intrauteriner Transmission.
Bis auf Thailand, wo sie die häufigste parasitäre Erkrankung des ZNS darstellt, ist sie jedoch selbst in ihren endemischen Gebieten (Japan, Korea, Laos, Malaysia, Taiwan, Thailand, Mexico, Ecuador) relativ selten.
FAZIT: ein gewiefter Parasit der nur mittels serologischem Test und Biopsien nachgewiesen werden kann. Vorsicht ist geboten bei "zu frischem" Fisch, dreckigem Wasser (Copepoden) und nicht garem Geflügel.
References:
[1] S. A. NADLER, R. A. CARRENO, H. MEJÍA-MADRID, J. ULLBERG, C. PAGAN, R. HOUSTON and J.-P. HUGOT (2007). Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology, 134 , pp 1421-1442
doi:10.1017/S0031182007002880
[2] Martina Wijova, Frantisek Moravec, Ales Horak, Julius Lukes, Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences, International Journal for Parasitology Volume 36, Issue 9, , August 2006, Pages 1067-1075.
doi:10.1017/S0031182007002880
[3] Moore DAJ, McCrodden J, DeKumyoy P, Chiodini PL. Gnathostomiasis: an emerging imported disease. Emerg Infect Dis [serial online] 2003 Jun
Besonders interessant ist die Gattung Gnathostoma tatsächlich durch ihre phylogenetische Position. Da ich zurzeizeit Hilfe von einem Diplomanden habe, der sich um die Phylogeny der Gattung Anguillicola und um eine bessere Auflösung des Baums für die basalen Spirurina kümmern soll, gibt es hier eine Neuheit- einen Gastpost "meines" Diplomanden Dominik Laetsch:
Grund für unser Interesse an der Gattung Gnathostoma ist also -wie gesagt- zum einen die geringe phylogenetische Distanz der SSU-rDNA-Sequenz dieses Nematoden zu jener unseres "Lieblings-Wurms" Anguillicola crassus und die Tatsache, dass die Familien Gnathostomatidae und Aguillicolidae aufgrund jener Daten den basalen Zweig der Spirurina darzustellen scheinen [1], [2]. Bestimmte Vertreter der Gattung Gnathostoma werden uns daher auch als "Out-group" in der phylogenetischen Analyse des Genus Anguillicola dienen (was das Thema meiner Diplomarbeit darstellt).
Der Genus Gnathostoma gliedert sich in ca. 9 mehr oder weniger gut von einander abgegrenzten Arten, welche wie alle Spirurina parasitisch leben. Davon sind mindestens drei als humanpathogene Arten beschrieben: G. turgidum, G. doloresi und G. spinigerum. Der Lebenszyklus von G. spinigerum ist bekannt und umfasst die Entwicklung der L1-Larve zum, für den Endwirt infektiösen, dritten Larvenstadium in zwei aquatischen Zwischenwirten (Copepoden des Genus Cyclops bzw. Fische/ Amphibia/ Mollusca) und die anschliessende Entwicklung zum Adultus im Endwirt (Mammalia; oft Feliden, Caniden und Suiden). Paratenische Wirte stellen vor allem Fische und Vögel dar. Im Endwirt durchbohrt G. spinigerum nach der oralen Aufnahme infizierten Gewebes die Magenwand und bereist als Larva migrans für die nächsten 3 Monate dessen Gewebe und innere Organ bis er zurückkehrt und sich an die mucosale Magenwand heftet. Dort entwickelt er sich für weitere 6 Monate bis er beginnt unembryonierter Eier zu produzieren, welche durch mit dem Kot ausgeschieden werden. Im Wasser embryonieren diese Eier, und mit der Azfnahme durch den ersten Zwischenwirt ist der Kreislauf geschlossen.
Das klinische Bild wird unter dem Begriff Gnathostomiasis [3] zusammengefasst und kann sowohl bei Feliden, Caniden als auch Hominiden letal verlaufen. Erstmals beschrieben wurde dieses Krankheitsbild 1835 anhand eines Kadavers eines jungen Tigers des Londoner Zoos. In Menschen entwickelt sich G. spinigerum nicht zum Adultus sondern migriert durch das Körpergewebe für bis zu 10- 12 Jahre, wobei zwischen kutaner und viszeraler Larva migrans unterschieden wird. Ersteres beschreibt nur Hautexzesse, bedingt durch mechanische Zerstörung des Gewebes und Produktion von Proteasen, Hyaluronidasen und Hemolysin durch den Parasiten, sowie durch die Immunreaktion des Wirtes. Letzteres umfasst die selben Faktoren jedoch in Organen wie der Leber und dem ZNS. Diese Scäden führen zu einer Mortalität von 8-25 % der betrofenen Patienten. Desweiteren gibt es drei dokumentierte Fälle von intrauteriner Transmission.
Bis auf Thailand, wo sie die häufigste parasitäre Erkrankung des ZNS darstellt, ist sie jedoch selbst in ihren endemischen Gebieten (Japan, Korea, Laos, Malaysia, Taiwan, Thailand, Mexico, Ecuador) relativ selten.
FAZIT: ein gewiefter Parasit der nur mittels serologischem Test und Biopsien nachgewiesen werden kann. Vorsicht ist geboten bei "zu frischem" Fisch, dreckigem Wasser (Copepoden) und nicht garem Geflügel.
References:
[1] S. A. NADLER, R. A. CARRENO, H. MEJÍA-MADRID, J. ULLBERG, C. PAGAN, R. HOUSTON and J.-P. HUGOT (2007). Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology, 134 , pp 1421-1442
doi:10.1017/S0031182007002880
[2] Martina Wijova, Frantisek Moravec, Ales Horak, Julius Lukes, Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences, International Journal for Parasitology Volume 36, Issue 9, , August 2006, Pages 1067-1075.
doi:10.1017/S0031182007002880
[3] Moore DAJ, McCrodden J, DeKumyoy P, Chiodini PL. Gnathostomiasis: an emerging imported disease. Emerg Infect Dis [serial online] 2003 Jun
Vergleichende Entwicklungsgenomik- Zwei Paper und massig Fehlinterpretationen
Evo-Devo (Evolutionary developmental biology) lässt sich am besten mit "Evolutionäre Entwicklungsbiologie" ins Deutsche übersetzen. Der Forschungszweig basiert darauf, dass natürliche Selektion oft nicht unmittelbar auf den Genotyp eines Individuums wirken kann, sondern auf den Phänotyp. Dieser Phänotyp wird durch ein komplexes genetisches Programm bei der Entwicklung des Organismus produziert. Bei einem solchen Entwicklungsprogramm sind die Mengen an Proteinen oder RNAs entscheidend; so können gewisse Schwellenwerte dieser Moleküle Zellschicksale beeinflussen und den Phänotyp bestimmen.
Eines der grundsätzlichsten Dogmen von Evo-Devo ist daher, dass Unterschiede in der Genexpression während der Entwicklung für unterschiedliche Phänotypen verantwortlich sind. Diese Expressionsunterschiede wiederum werden durch Unterschiede in nicht kodierenden, den Genen vorgelagerten (cis-)regulatorischen Sequenzen (hauptsächlich Promotoren und Enhancer) oder in den Protein-Sequenzen von übergeordneten Transkriptionsfaktoren verursacht (oder wiederum in der Expression dieser Transkriptionsfaktoren).
Evo-Devo will daher in die bisweilen postulierte Lücke zwischen Macro- und Microevolution vorstoßen. Gerade Experten auf dem Gebiet der Entwicklungsbiologie räumen mitunter die Möglichkeiten saltatorischer Evolution ein. Das heißt, sie halten Makromutationen für möglich, die in einem einzigen Mutationsschritt starke Änderungen des Phänotyps (sogenannte "Hopeful Monster") produzieren. Dies soll hauptsächlich durch Änderungen an wichtigen "Schaltstellen"-Transkriptinsfaktoren in den Entwicklungsprogrammen geschehen. Diese Sichtweise wird von der Mehrzahl der Evolutionsbiologen -bis auf Darwin selbst zurückgehend- abgelehnt. Die konventionelle Theorie besagt, dass Veränderungen durch graduelle Mutationen erfolgen, die sich über Generationen akkumulieren. Einzelne Wissenschaftler auf dem Gebiet der evolutionären Entwicklungsbiologie versuchen also mitunter Modifikationen an den bestehenden Fundamenten der Evolutionsbiolgie zu erreichen, was auf heftige Kritik (auch aus den eigenen Reihen) stößt. Wahrscheinlich ist Evo-Devo daher eines der lebendigsten Forschungsgebiete in der aktuellen Biologie.
Die von mir im Folgenden besprochenen Studie untersuchen beide dieses zentrale Dogma von Evo-Devo genauer und lassen meines Erachtens auch Schlüsse zur genannten Kontroverse zu.
Cretekos et al. benutzen dazu die Unterschiede im Wachstum der Vorderextremitäten zwischen Carollia perspicillata (einer Fledermaus) und der Maus. Beide Taxa stammen aus unterschiedlichen Ordnungen der Säugetiere Chiroptera (=Fledertiere) beziehungsweise Rodentia (=Nagetiere), teilen also einen gemeinsamen Vorfahren vor etwa 80-100 Millionen Jahren.
Die Studie betrachtet Veränderungen in Prx1, einem Transkriptionsfaktor, der durch klassische entwicklungsbiologische Methoden als wichtig in der Extremitätenentwicklung identifiziert wurde. Die Kollegen stellten beim Verglich der Gen-Sequenzen aus Maus und Fledermaus nur einen nicht-synonymen (in das Protein übersetzten)Unterscheid fest. Dieser Unterschied befindet sich in einem Bereich des Genes, der ohnehin wenig konserviert ist, und nicht mit der typischen Funktion des Traskriptionsfaktors in Verbindung gebracht wird.
Unterschiede in der Expression von Prx1 konnten in der späten Entwicklung der Vorderextremitäten festgestellt werden, in der Fledermaus war der Transkriptionsfaktor speziell im Bereich der Handwurzelknochen stärker exprimiert. Dieses Ergebnis korreliert gut mit dem zu diesem Zeitpunkt verstärkt auftretenden Längenwachstum in der Fledermaus.
Cretekos et al. betrachteten weiterhin also einen Enhancer "stromaufwärts" des Gens, der ebenfalls mit klassischen Methoden identifiziert worden war. Dieser Enhancer enthält zwei Bereiche die zwischen Nager und Fledertier relativ konserviert sind, in diesen wurde dann die Funktion vermutet. Um dies zu testen konnte die Gruppe die jeweilige Enhancer-Region an ein Reportegen koppeln und in Mäuse einbringen, dabei wurde stärkere Reporter-expression beim Chiroptera-Enhancer beobachtet.
Doch damit nicht genug, der Gruppe gelang es schließlich das Fledermaus-Kontrollelement in die Maus einzubringen, so dass es die Expression des Prx1-Gens steuert. Die entsprechenden Mäuse zeigten tatsächlich ein verstärktes Wachstum der Vorderextremitäten.
Die Studie konnte so eindrucksvoll das zentrale Dogma der evolutionären Entwicklungsbiologie bestätigen und weiterhin zeigen, dass die zugrunde liegenden Veränderungen im untersuchten Fall auf Enhancer-Elementen basieren.
Auch die Studie von Prabhakar et al. beschäftigt sich mit der Funktion von Enhancern, die betreffende genomische Region war aber mit anderen Mitteln identifiziert worden. Dabei wurden komplett sequenzierten Genome von Wirbeltieren nach konservierten, nichtkodierenden Sequenzen durchsucht. Aus diesen Sequenze wurden wiederum jene identifiziert, die in der Menschlichen Linie (entgegen des allgemeinen Trends) evolvieren. Diese Vorgehen basiert auf der Tatsache, dass Elemente mit einer bestimmten Funktion weniger evolvieren als funktionslose Elemente, ändern sie allerdings ihre Funktion erfolgt die Evolution sogar schneller als dies unter Neutralität (Funktionslosigkeit) der Fall wären. Die identifizierte Region liegt im Intron eines Gens, das mit der Funktion des Endosoms in Verbindung gebracht wird. Weier "stromäbwärts" befindet sich wieder ein Transkriptionsfaktor, dessen Wirkung die Entwicklung der Gliedmaßen beeinflusst. Die Expression welcher Gene genau der mögliche Enhancer beeinflusst ist also noch nicht geklärt.
Zur Untersuchung der Funktion der Enhancer-Region benutzten die Kollegen also wieder ein Reporterassay (ß-Galactosidase). Sie brachten die postulierten Kontrollelemente aus Rhesusaffen (Macaca mulatta), Schimpansen (Pan troglodytes) und dem Menschen (Homo sapiens) (Divergenz vor 6 bzw. 25 Mya) gekoppelt an das Reportergen, in Mäuse ein. So konnten die Forscher zeigen, dass die menschlichen Elemente im Vergleich zu denen aus beiden anderen Primaten, verstärkt Gene beim Wachstum der Extremitäten anschalten. Sie konnten so die durch den Genom-Verglich identifizierten Unterschiede bestätigen: Der Zustand und die Wirkung des Enhancers sind sich in nicht-menschlichen Affen ähnlich und entsprechen daher wahrscheinlich dem Zustand im gemeinsamen Vorfahren.
Mit sehr cleveren Experimenten bewiesen die Kollegen, dass genau die 13 Basen Unterschied im Menschen im Vergleich zu Schimpanse und Rhesusaffe den Unterschied ausmachen. Sie konstruierten Fragmente aus den beiden "Vierbeinern" in denen nur die betroffenen 13 Unterschiede eingebracht waren. Diese Konstrukte hatten die gleiche Wirkung wie das original-menschliche Element.
Die Studie konnte so, aufbauend auf dem zuvor beschriebenen zentralen Dogma von Evo-Devo, zeigen, dass durch Genomvergleiche die betreffenden Elemente identifiziert werden können. Ein denkbarer Name für ein solches Vorgehen ist "Vergleichende Entwiklungsgenomik" oder im Englischen "Comparative developmental genomics".
Weiter demonstrieren beide Studien eindrucksvoll die evolutionären Möglichkeiten für graduelle Veränderungen in Entwicklungsprogrammen. In beiden Beispielen konnte durch Veränderung einzelner Basen in regulativen Bereichen Unterschiede in der Genexpression erzeugt werden. Da es sich um Variationen in einzelnen Basenpaaren (SNPs) handelt, die langsam und nacheinander ins Genom einfließen, verlangen die hier beobachteten Unterschiede geradezu nach Gradualismus.
Fee hat vor einigen Wochen auf dem Blog Science-meets-spciety ebenfall über das letztere Paper geschrieben und ich möchte einige Fehler in diesem (auch auf Researchblogging erschienenen) Post abschließend korrigieren. Ich hoffe so eine Diskussion anzuregen, falls es im deutschsprachigen Raum genügend Interesse gibt.
S-M-S:
Wir teilen bis zu 98% der codierenden DNA mit unseren nächsten Verwandten, den Schimpansen (Pan troglodytes) und doch unterscheiden wir uns markant von ihnen.
Das stimmt nicht! Eine der grundlegendsten Entdeckungen der letzten Jahre waren Unterschiede in der Kopienzahl einzelner Gene im Menschlichen Genom. 2007 wurde so (Hauptsächlich durch den Vergleich der Genome von Venter und Watson) offensichtlich, dass einzelne Menschen sich in 2-3% ihres Genoms unterscheiden. Der Unterschied zu unseren nächsten interspeziefschen Verwandten dürfe daher mindestens 5% betragen.
S-M-S:
Der größte Teil der DNA in menschlichen Zellen ist nicht kodierend und wurde, als man dies entdeckte, fälschlicherweise als Junk-DNA (Abfall-DNA) verschrien.
Diese Sichtweise ist grundsätzlich falsch! Es wurden seit der Entdeckung nicht-kodierender Bereiche schon versucht Funktionen für diese für eine höhere Organisation zu postulieren. Vergleicht man aber unterschiedliche Organismen (z.B. der Zwiebel oder des Salamanders) mit der des Menschen oder der Kugelfische (Tetraodontidae; anderes Extrem) fällt schnell auf dass eine höhere Komplexität nicht mit der Genomgröße korreliert.
Die in Frage stehenden regulatorischen Elemente machen einen winzigen Teil des Genoms aus.
S-M-S:
Dabei stiessen sie auf einen Abschnitt von 546 Basenpaaren Länge, der sich seit der Entwicklung der Wirbeltiere nur wenig verändert hatte. Jedoch hatten sich in der relativ kurzen Zeit von 6 Millionen Jahren, seit sich die Entwicklungszweige von Mensch und Schimanse trennten, 16 Veränderungen etabliert, die alle in einem Abschnitt von 81 Basenpaaren clusterten. So etwas ist für einen genetischen Detektiv ein eindeutiges Indiz, dass weitere Untersuchungen gewinnbringend sein könnten.
Die Forscher wussten im Gegensatz zum Schreiber dieser Zeilen um die Existenz von Enhancen. Andernfalls hätten sie den entsprechenden Bereich nicht als solchen identifizieren können. In der besprochenen Studie wurden nicht zum ersten Mal Selektion auf einen nicht-kodierenden Bereich nachgewiesen. Ähnliche Fehlinterpretationen und die übertriebene Darstellung von Neuheiten (wissenschaftlichen Revolutionen) in der Wissenschaftsberichterstattung veranlassen auch beispielsweise Kreationisten regelmäßig zu ähnlichen Dummheiten.
S-M-S:
[...]so aktivierten alle die Expression von Genen in den Augen, Ohren und in den embryonalen Kiemenbögen, die später den Kiefer bilden.
Die Expression eines Reportergens! Dies ist im Vergleich zu den anderen Fehlern aber eher zweitrangig.
S-M-S:
Ein neuer Teilbereich der entwicklungsgenetischen Forschung, der bestimmt noch viele Überraschungen und neue Erkenntnisse birg.
Quatsch! "Vergleichende Entwicklungsgenomik" ist zwar ein recht neuer boomender Bereich der Entwicklunsbiologie, erfunden wurde er aber in diesem Paper nicht. Die Studie bestätigt vielmehr experimentell die Validität der zugrundeliegenden in silico Analysen.
Ich hab hier mal die jährliche Zähl an Veröffentlichungen, die "Comparative developmental genomics" im Volltext (Pub-med) erwähnen geplotet:
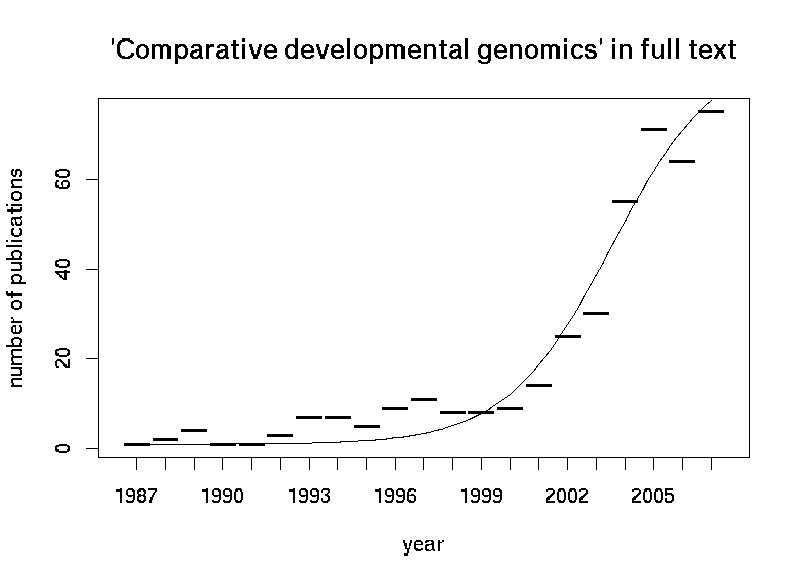
Dabei fällt auf, dass das erste Paper bereits aus dem Jahre 1988 stammt, als eigentlich noch keine Genome zum Vergleich bereitstanden. Veröffentlichungen vor dem Jahre 2001 können also wahrscheinlich als "Hintergrundrauschen" oder "Vorahnung" interpretiert werden, wirkliche "Comparative genomics" in dem heute etablierten Sinn waren damals noch kaum möglich. Ab diesem Zeitpunkt wurden dann anhand der vorhandenen Daten die betreffenden Methoden entwickelt. Prabhakar et al. haben also zwar eine schöne Studie angefertigt, mitnichten aber das "Teilgebiet" erfunden.
Kommentare/ Kritik erwünscht, ich bin kein Entwicklungsbiologe und habe sicher selbst Fehler gemacht!
________________________________________________________________________________________________________________
C. J. Cretekos, Y. Wang, E. D. Green, J. F. Martin, J. J. Rasweiler, R. R. Behringer (2008). Regulatory divergence modifies limb length between mammals Genes & Development, 22 (2), 141-151 DOI: 10.1101/gad.1620408
S. Prabhakar, A. Visel, J. A. Akiyama, M. Shoukry, K. D. Lewis, A. Holt, I. Plajzer-Frick, H. Morrison, D. R. FitzPatrick, V. Afzal, L. A. Pennacchio, E. M. Rubin, J. P. Noonan (2008). Human-Specific Gain of Function in a Developmental Enhancer Science, 321 (5894), 1346-1350 DOI: 10.1126/science.1159974
Freitag, 26. September 2008
Nochmal Aua: E.O. Wilson in der NYTimes
Schämen muss man sich eigentlich nicht wenn, man so nen Fehler macht. Fee ist in guter Gesellschaft, wie ich in diesem Post auf Myrmecos erfahren habe:
;-)
Given the antiquity of the lineage, the temptation to view Martialis as an ur-ant of sorts is strong. E.O. Wilson certainly felt that way when interviewed for a recent NYTimes article:With due respect to Wilson, such a view is a mistake. Martialis has over 120 million years’ separation since the ur-ant, plenty of time to develop along its own trajectory.Dr. Wilson…is trying to contain his excitement: the 14,001st ant species has just been discovered in the soils of a Brazilian forest. He steamrolls any incipient skepticism about the ant’s uniqueness — the new species is a living coelacanth of ants, a primitive throwback to the first ant, a wasp that shed its wings and assigned all its descendants to live in earth, not their ancestral air. The new ant is so alien, Dr. Wilson explains, so unlike any known to earthlings, that it will be named as if it came from another planet.
;-)
Dienstag, 23. September 2008
Keine Diskussion erwünscht?
Ein Post aus unerfreulichem Anlass... zunächst die Vorfälle in chronologischer Abfolge ohne Bewertung.
Durch den deutschen Arm von Research-Blogging bin ich Ende letzter Woche auf einen Post auf dem Blog Science-meets-Society aufmerksam geworden. Fee und Uli schreiben dort auf leicht verständliche Art über ein sehr breites Spektrum wissenschaftlicher Veröffentlichungen.
Bei einem kurzen Blick auf nur einen Post über den Fund einer neuen Ameisengattung (Paper) ist mir dann aber ein gewaltiger Fehler aufgefallen:
Mein erster Kommentar (genau wiedergeben kann ich diesen nicht, dazu später mehr) war aus Zeitmangel etwas barsch:
Meine Meinung dazu:
Im deutschsprachigen Raum fehlt bislang eine Diskussionskultur auf Wissenschaftsblogs. Researchblogging ist sicher ein guter Ansatz dies zu ändern. Kritik an Blogposts muss auch in ungeschminkter Form erlaubt sein (dieser Post ist übrigens genauso grottenschlecht, und wird von Argent23 "mit Samthandschuhen" kritisiert). Der Name des Blogs (Science-meets-society; science communication) ist im Zusammenhang mit der Löschung unerwünschter Kommentare geradezu lächerlich.
Ich bin ein großer Fan guter Vereinfachungen von komplexen Zusammenhängen in populärwissenschaftlichen Texten zur Evolutionsbiologie und allgemein von Wissenschaftsjournalismus. Allerdings braucht man um den betreffenden Sprachgebrauch zu beherrschen eine fundierte Kenntnis des Fachgebiets und einige Erfahrung (wegen letzterem lesen sich manche meiner Posts vielleicht auch etwas kryptisch). Gerade die Vereinfachung macht die Sache eben nicht leichter.
Zum thematischen Ursprung des Disputs noch soviel: Wer sich weiter informieren möchte kann dies auf deutschen Blogs noch nicht (falls doch bin ich dankbar für nen Link). Ich werd mich drum kümmern sobald ich etwas mehr Zeit hab! Über Fallstricke im Verständnis von Evolutionsbiologie kann man sich solange super (besser als ich das wahrscheinlich jemals hinbekommen werde) bei T. Tyan Gregory informieren.
Durch den deutschen Arm von Research-Blogging bin ich Ende letzter Woche auf einen Post auf dem Blog Science-meets-Society aufmerksam geworden. Fee und Uli schreiben dort auf leicht verständliche Art über ein sehr breites Spektrum wissenschaftlicher Veröffentlichungen.
Bei einem kurzen Blick auf nur einen Post über den Fund einer neuen Ameisengattung (Paper) ist mir dann aber ein gewaltiger Fehler aufgefallen:
Phylogenetische Analysen haben nun gezeigt, dass es sich bei Martialis wahrscheinlich um den ältesten bekannten Vorfahren der modernen Ameisen handelt, der in einem so stabilen Lebensraum wie dem Regenwaldboden, lange Zeit überdauern konnte. Dies wirft ein völlig neues Licht auf die Evolution und die Entwicklung von außergewöhnlichen Lebensweisen, heute bekannter Ameisen.
Mein erster Kommentar (genau wiedergeben kann ich diesen nicht, dazu später mehr) war aus Zeitmangel etwas barsch:
Aua! Fehler! Eine moderne (heute lebende) Ameisengattung kann niemals der Vorfahre aller heute lebender Ameisengattungen sein.Nach Lesen des Papers hab ich dann etwa folgendes geschrieben:
Die Autoren lehnen sich im "absract" weit aus dem Fenster indem sie- trotz sehr dürftiger Hinweise- spekulieren, die gefundene Ameisenart könnte ihrem mit allen anderen Ameisen gemeinsamen Vorfahren sehr ähnliche sein. In den "conclusions" relativieren sie dann allerdings ihre Hypothese wieder; " The exact nature of the ancestral ant remains uncertain given that the propensity for repeated evolution of an epigaeic lifecycle".Überraschenderweise waren diese beiden Kommentare Tags darauf aus dem Blog verschwunden.Seit dem habe ich zweimal versucht Fee und Uli zu kontaktieren, leider aber keine Antwort erhalten. Ein weiterer Kommentar wurde gelöscht.
Ähnliche Fehler wurden in der populärwissenschaftlichen Berichterstattung über das Platypus-Genom-Paper haufenweise gemacht und un z.B. von T.Ryan Gregory sehr gut diskutiert.
Meine Meinung dazu:
Im deutschsprachigen Raum fehlt bislang eine Diskussionskultur auf Wissenschaftsblogs. Researchblogging ist sicher ein guter Ansatz dies zu ändern. Kritik an Blogposts muss auch in ungeschminkter Form erlaubt sein (dieser Post ist übrigens genauso grottenschlecht, und wird von Argent23 "mit Samthandschuhen" kritisiert). Der Name des Blogs (Science-meets-society; science communication) ist im Zusammenhang mit der Löschung unerwünschter Kommentare geradezu lächerlich.
Ich bin ein großer Fan guter Vereinfachungen von komplexen Zusammenhängen in populärwissenschaftlichen Texten zur Evolutionsbiologie und allgemein von Wissenschaftsjournalismus. Allerdings braucht man um den betreffenden Sprachgebrauch zu beherrschen eine fundierte Kenntnis des Fachgebiets und einige Erfahrung (wegen letzterem lesen sich manche meiner Posts vielleicht auch etwas kryptisch). Gerade die Vereinfachung macht die Sache eben nicht leichter.
Zum thematischen Ursprung des Disputs noch soviel: Wer sich weiter informieren möchte kann dies auf deutschen Blogs noch nicht (falls doch bin ich dankbar für nen Link). Ich werd mich drum kümmern sobald ich etwas mehr Zeit hab! Über Fallstricke im Verständnis von Evolutionsbiologie kann man sich solange super (besser als ich das wahrscheinlich jemals hinbekommen werde) bei T. Tyan Gregory informieren.
Samstag, 20. September 2008
Powerful statistics
Jeder, der schonmal gedacht hat er hätte in einer Präsentation chronologische Daten optimal visualisiert sollte sich das hier mal anschauen...
Hans Rosling bei einem TED-Vortrag
Wie geil ist da eigentlich? Die Idee dahinter nennt sich Gapminder und ist als Google Gadget verfügbar. Jetzt brauch ich nur noch Wurm-Daten, die so ne Darstellung erlauben :-).
Hans Rosling bei einem TED-Vortrag
Wie geil ist da eigentlich? Die Idee dahinter nennt sich Gapminder und ist als Google Gadget verfügbar. Jetzt brauch ich nur noch Wurm-Daten, die so ne Darstellung erlauben :-).
Freitag, 19. September 2008
Montag, 8. September 2008
Würmer in Paris: Compatibility polymorphism in snail/trematode interactions
Die Interaktion von Trematoden und Schnecken beruht (nach der aktuellen Hypothese) darauf, dass das sich zur Muttersporocyste entwickelnde Miracidium des Parasiten den Wirt entweder aktiv in seiner Immunantwort beeinflusst, oder aber durch ein Mimikry von Wirtsepitopen vom Immunsystem unerkannt bleibt. Letzteres ist bei B. glabrata und S. mansoni der Fall.
Während bei einer Beeinflussung/Unterdrückung des Immunsystems in anderen Wirt/Parasit-Systemen generell von anfälligen und resistenten Wirten gesprochen werden kann, ist die Interaktion beim Mimikry von Wirtsepitopen komplizierter:
Die selbe Parasiten-Linie ist unterschiedlich kompatibel mit künstlich auf Resistenz oder Anfälligkeit selektierten Schnecken-Linien (Soweit wäre das natürlich auch durch bloße Resistenz der Schnecke erklärbar). Werden aber anderer Schnecken-Linien mit einer anderen Parasiten-Linie selektiert ist die in dieser künstlichen Co-Evolution resistente Schneckenlinie anfällig für die Parasiten-Linie aus der ersten Selektion. Ein schönes Schaubild dazu (Fig.3) kann man sich in diesem Paper anschauen, wenn man Zugriff hat.
Die Versuche der Gruppe zur Identifizierung der molekularen Grundlage dieser Interaktion wurden nun mit einem zu einer brasilianischen Schnecken-Linie kompatiblen Stamm von S. mansoni und einem inkompatiblen Stamm durchgeführt (Zur Vermehrung des inkompatiblen Stamms stand eine andere Schneckenlinie zur Verfügung).
Verwendet wurde ein proteomischer Ansatz (2D Gele) und es konnten sogenannte Schistosoma mansoni polymorphic mucin-like proteins (Sm PoMuc) als Hauptverdächtige identifiziert werden.
Mucine oder Mucin-ähnliche Proteine sind im Schleim (Mucus) vieler Organismen enthalten. Solcher Mucus wird oft von als Reaktion auf eine Infektion produziert, und kann dazu dienen Parasiten zu bekämpfen. Andererseits sekretieren Parasiten aber auch Mucin-ähnliche Moleküle um die Wirtsabwehr zu täuschen.
In Sm PoMucs werden fast ausschließlich in den in der Schnecke parasitierenden Stadien exprimiert. In den Miracidien werden sie in sogenannten Apicaldrüsen produziert und sekretiert. Möglicherweise kann sich der Parasit so in einem küstlichen Nebelschleier verbergen (im Deutschen klingt das schon fast poetisch; anders als das Englische "covered by a smoke screen").
Die Struktur der Sm PoMucs erwies sich als höchst polymorph. Gemeinsam haben alle Transkripte ein Signalpeptid (in Übereinstimmung mit der extrazellulären Lokalisation) gefolgt von einer Region bestehend aus einer variablen Anzahl von (3 leicht unterschiedlichen; r1, r1' und r2) 9-Aminosäuren-Repeats gefolgt von 3 leicht unterschiedlichen C-terminalen Regionen (1, 2, 3).
Eine unterschiedliche Kombination dieser Elemente kennzeichnet drei Gruppen von Sm PoMucs (die erste Gruppe und die zweite Gruppe haben nur r2 und unterschiedliche C-terminale Regionen 1 und 2; die dritte Gruppe hat R1' und R1 gefolgt von der C-terminalen Region 3).
Eine vierte Gruppe wiest eine wahrscheinlich durch alternatives Splicen noch stärker abweichende Sequenz auf.
Im Kompatiblen und inkompatiblen Parasiten-Stamm unterscheidet sich in allen drei Gruppen die Anzahl der Repeats. Codiert werden die drei Gruppen von jewels einem Gen. Die unterschiedliche Anzahl der Repeats entsteht ebenfalls durch alternatives splicen.
Besonders von Bedeutung ist weiterhin, das die Repeats Serin-, Threonin- und Prolin-reich sind, was als Zeichen für eine posttranslationale Glycolsylierung gilt.
So könnten möglicherweise speziell die unterschiedlich angefügten Zucker für die unterschiedlichen Eigenschaften der Moleküle in der Wirts-Parasit-Interaktion verantwortlich sein.
Für mich war der Vortrag deshalb so interessant, da er mir die Beschränkungen der in meinem Projekt verwendeten transkriptomischen Methoden vor Augen geführt hat:
Allein das Assembley der Repeats wäre aus 454-Pyrosequenz-Daten (bei der bisherigen read-Länge von 250 Basen) wahrscheinlich sehr schwierig bis unmöglich. Und selbst mit sehr viel längeren reads bräuchte man eine extrem hohe Coverage der betreffenden Regionen um ein solches Maß an Polymorphismus aufdecken zu können.
Außerdem interessant (und etwas beunruhigend, wenn man die Arbeit mit Proteinen nicht besonders mag) ist die Tatsache, dass ein wesentlicher Teil des Polymorphismus durch posttranslationale Modifikation entstehen kann. Generell neu und überraschend ist das natürlich nicht unbedingt, allerdings könnte es besonders im Co-Evolutions-Context eine gewaltige Rolle spielen.
Gerade in der Interaktion mit mehreren sympatrischen Wirtsarten könnten solche Mechanismen zu einer Evolution von Plastizität führen. Speziell diese Plastizität könnte als Gegenspieler von adaptiven Prozessen sympatrische, ökologische Artbildung von den an unterschiedliche Wirtsorganismen angepassten Parasiten-Stämmen verhindern.
_______________________________________________________________________________________________________________________________________
E ROGER, B GOURBAL, C GRUNAU, R PIERCE, R GALINIER, G MITTA (2008). Expression analysis of highly polymorphic mucin proteins (Sm PoMuc) from the parasite Schistosoma mansoni☆ Molecular and Biochemical Parasitology, 157 (2), 217-227 DOI: 10.1016/j.molbiopara.2007.11.015
Samstag, 6. September 2008
Würmer in Paris: Ist DNA-barcoding unfair?
Ein Großteil der Diskussion nach dem DNA-barcoding Symposium auf dem EMOP hatte anstatt der technischen Schwierigkeiten [1] leider eine andere Grundlage:
3. R. Guerrero stellte die Schwierigkeiten der Forscher aus "Schwellenländern" sich umfangreiche Sequenzierungen finanziell leisten zu können in den Vordergrund:
Dadurch würden gerade in diesen Ländern, in denen die Biodiversität am größten ist keine Fortschritte erzielt, während in den entwickelten Ländern, mit fast vollständig beschriebener Diversität nach kryptischen Arten gesucht wird.
Ganz von der Hand zu weißen sind diese Einwände wahrscheinlich nicht, da selbst wenn Sequenzierungen immer günstiger werden, das Anlegen von großen Datenbanken, die Morphologie (Museumsexemplare, Bilder, Videos), ökologische Parameter (Verbreitung) mit den Sequenzdaten verknüpfen immer noch teuer bleibt.
Andererseits: Kein Doktorrand wird sich dagegen sträuben im Rahmen seiner Dissertation in die Tropen zu reisen um umfangreiche Proben zu nehmen, die dann daheim sequenziert werden. Und auch der durchschnittliche Arbeitsgruppenleiter auf seinem speziellen Gebiet wird (sobald Hochdurchsatzsequenzierung mal wirklich nichts mehr kostet und weit verbreitet ist) den Vorteil und das Prestige erkennen den die Entdeckung hunderter "möglicher Arten" bietet.
[1] Davon gibt es (scheinbar?) genügend: Alleine F1000 listet 3 Paper, die von Problemen berichten (in Fällen, wo die intraspezifische Varianz die interspezifische überlappt) und 3 Paper die Erfolge beschreiben.
Zurzeit gibt es außerdem ein viel diskutiertes Paper über Probleme durch COI-Pseudogene im Kern...
...das ganze ist schwer zu beurteilen und natürlich spielen dabei die persönlichen Arbeitsweisen eine Rolle. Ein deutscher Evolutionsökolge meinte scherzhaft zu einem Kollegen, als ich ihn darauf ansprach dass ich gerne alle Aalparasiten barcoden würde: "Vorsicht, der will uns wegrationalisieren: Dann schmeisst er nur noch alles in nen Mixer und zählt Sequenzen..."
3. R. Guerrero stellte die Schwierigkeiten der Forscher aus "Schwellenländern" sich umfangreiche Sequenzierungen finanziell leisten zu können in den Vordergrund:
Dadurch würden gerade in diesen Ländern, in denen die Biodiversität am größten ist keine Fortschritte erzielt, während in den entwickelten Ländern, mit fast vollständig beschriebener Diversität nach kryptischen Arten gesucht wird.
Ganz von der Hand zu weißen sind diese Einwände wahrscheinlich nicht, da selbst wenn Sequenzierungen immer günstiger werden, das Anlegen von großen Datenbanken, die Morphologie (Museumsexemplare, Bilder, Videos), ökologische Parameter (Verbreitung) mit den Sequenzdaten verknüpfen immer noch teuer bleibt.
Andererseits: Kein Doktorrand wird sich dagegen sträuben im Rahmen seiner Dissertation in die Tropen zu reisen um umfangreiche Proben zu nehmen, die dann daheim sequenziert werden. Und auch der durchschnittliche Arbeitsgruppenleiter auf seinem speziellen Gebiet wird (sobald Hochdurchsatzsequenzierung mal wirklich nichts mehr kostet und weit verbreitet ist) den Vorteil und das Prestige erkennen den die Entdeckung hunderter "möglicher Arten" bietet.
[1] Davon gibt es (scheinbar?) genügend: Alleine F1000 listet 3 Paper, die von Problemen berichten (in Fällen, wo die intraspezifische Varianz die interspezifische überlappt) und 3 Paper die Erfolge beschreiben.
Zurzeit gibt es außerdem ein viel diskutiertes Paper über Probleme durch COI-Pseudogene im Kern...
...das ganze ist schwer zu beurteilen und natürlich spielen dabei die persönlichen Arbeitsweisen eine Rolle. Ein deutscher Evolutionsökolge meinte scherzhaft zu einem Kollegen, als ich ihn darauf ansprach dass ich gerne alle Aalparasiten barcoden würde: "Vorsicht, der will uns wegrationalisieren: Dann schmeisst er nur noch alles in nen Mixer und zählt Sequenzen..."
Würmer in Paris: Acoel Flatworms
2. Tim Littlewood brachte den besten Witz während eines Vortrags und schloss gleichzeitig eine Lücke in meinem Wissen über Phylogenie der Protostomier:
Er erwähnte, dass sehr viele lateinische Taxa-bezeichnungen in verschiedenen Sprachen völlig unterschiedlich ausgesprochen würden. So würde "Acoel" schon im deutschen und englischen etwas unterschiedlich ausgesprochen, die Franzosen sprächen es sogar "ashole" aus. Originalzitat: "but dont be a ashole (acoel) try to understand each other"...
Was hat es nun mit diesen (nicht-parasitischen) Würmern auf sich?
Wie man als halbwegs gebildeter Biologie (und erst recht als Zoologe im weitesten Sinne) wissen sollte hat die Phylogenie der Tiere in den letzen zehn Jahren [1] eine Revision erfahren [2]:
Dabei gibt es zwei große Kladen innerhalb der bilateralsymmetrischen Tiere, die Protostomier und die Deuterostomier (zu denen wir Vertebraten gehören).
Die Protostomier enthalten wiederum zwei Kladen: Eine die alle Tiere mit "sich häutenden" (moulting) Larvenstadien (Nematoden, Cheliceraten, etc.) umfasst und Ectysozoa genannt wird und eine zweite die Lophotrochozoa genannt wird.
Der Name Lophotrochozoa ist eine Neuschöpfung aus den Namen der beiden alten Kladen Trochozoa und Lophophorata.
Diese Lophotrochozoa enthalten neben den prominenten Mollusken und Anneliden auch die Platyhelminthen (hauptsächlich Trematoden und Cestoden für die Parasitologen). Dise alte Klade erwies sich aber eben auch als polyphyletisch und die "Acoel Flatworms" mussten ausgegliedert und an eine basale Position [3] als Schwestergruppe der übrigen Bilateria [4] gestellt werden.
Quellen im Text verlinkt!
Wobei ich jeweils zu der genererellen Revision [1+2] und den "Acoel Flatworms" [3+4] DAS klassische und ein neues Paper mit einer phylogenomischen Analyse verlinkt hab. (Phylogenomik bezieht sich hier auf die Analyse mehrerer (vieler) Genen aus großen EST-Datensätzen, nicht auf Jonathan Eisens Definition.)
Er erwähnte, dass sehr viele lateinische Taxa-bezeichnungen in verschiedenen Sprachen völlig unterschiedlich ausgesprochen würden. So würde "Acoel" schon im deutschen und englischen etwas unterschiedlich ausgesprochen, die Franzosen sprächen es sogar "ashole" aus. Originalzitat: "but dont be a ashole (acoel) try to understand each other"...
Was hat es nun mit diesen (nicht-parasitischen) Würmern auf sich?
Wie man als halbwegs gebildeter Biologie (und erst recht als Zoologe im weitesten Sinne) wissen sollte hat die Phylogenie der Tiere in den letzen zehn Jahren [1] eine Revision erfahren [2]:
Dabei gibt es zwei große Kladen innerhalb der bilateralsymmetrischen Tiere, die Protostomier und die Deuterostomier (zu denen wir Vertebraten gehören).
Die Protostomier enthalten wiederum zwei Kladen: Eine die alle Tiere mit "sich häutenden" (moulting) Larvenstadien (Nematoden, Cheliceraten, etc.) umfasst und Ectysozoa genannt wird und eine zweite die Lophotrochozoa genannt wird.
Der Name Lophotrochozoa ist eine Neuschöpfung aus den Namen der beiden alten Kladen Trochozoa und Lophophorata.
Diese Lophotrochozoa enthalten neben den prominenten Mollusken und Anneliden auch die Platyhelminthen (hauptsächlich Trematoden und Cestoden für die Parasitologen). Dise alte Klade erwies sich aber eben auch als polyphyletisch und die "Acoel Flatworms" mussten ausgegliedert und an eine basale Position [3] als Schwestergruppe der übrigen Bilateria [4] gestellt werden.
Quellen im Text verlinkt!
Wobei ich jeweils zu der genererellen Revision [1+2] und den "Acoel Flatworms" [3+4] DAS klassische und ein neues Paper mit einer phylogenomischen Analyse verlinkt hab. (Phylogenomik bezieht sich hier auf die Analyse mehrerer (vieler) Genen aus großen EST-Datensätzen, nicht auf Jonathan Eisens Definition.)
Abonnieren
Posts (Atom)
