
In diesem Post möchte ich anhand zweier Papern diskutieren, was Evo-Devo im allgemeinen ist und eine Unterdisziplin, die man im Deutschen wohl am besten "Vergleichende Entwicklungsgenomik" nennt, vorstellen. Außerdem möchte ich zeigen, warum Arbeiten auf diesem Gebiet für unser Verständnis der Evolutionstheorie wichtig sind. Zum Schluss möchte ich noch einen verwandten Beitrag auf dem deutschen
Researchblogging kritisieren und zeigen warum dieser Fehlinterpretationen und schlichte Fehlinformation enthält.
Evo-Devo (Evolutionary developmental biology) lässt sich am besten mit "Evolutionäre Entwicklungsbiologie" ins Deutsche übersetzen. Der Forschungszweig basiert darauf, dass natürliche Selektion oft nicht unmittelbar auf den Genotyp eines Individuums wirken kann, sondern auf den Phänotyp. Dieser Phänotyp wird durch ein komplexes genetisches Programm bei der
Entwicklung des Organismus produziert. Bei einem solchen Entwicklungsprogramm sind die Mengen an Proteinen oder RNAs entscheidend; so können gewisse Schwellenwerte dieser Moleküle Zellschicksale beeinflussen und den Phänotyp bestimmen.
Eines der grundsätzlichsten Dogmen von Evo-Devo ist daher, dass Unterschiede in der Genexpression während der Entwicklung für unterschiedliche Phänotypen verantwortlich sind. Diese Expressionsunterschiede wiederum werden durch Unterschiede in nicht kodierenden, den Genen vorgelagerten (cis-)regulatorischen Sequenzen (hauptsächlich
Promotoren und
Enhancer) oder in den Protein-Sequenzen von übergeordneten
Transkriptionsfaktoren verursacht (oder wiederum in der Expression dieser Transkriptionsfaktoren).
Evo-Devo will daher in die bisweilen postulierte Lücke zwischen Macro- und Microevolution vorstoßen. Gerade Experten auf dem Gebiet der Entwicklungsbiologie
räumen mitunter die Möglichkeiten saltatorischer Evolution ein. Das heißt, sie halten Makromutationen für möglich, die in einem einzigen Mutationsschritt starke Änderungen des Phänotyps (sogenannte "Hopeful Monster") produzieren. Dies soll hauptsächlich durch Änderungen an wichtigen "Schaltstellen"-Transkriptinsfaktoren in den Entwicklungsprogrammen geschehen. Diese Sichtweise wird von der Mehrzahl der Evolutionsbiologen -bis auf Darwin selbst zurückgehend-
abgelehnt. Die konventionelle Theorie besagt,
dass Veränderungen durch graduelle Mutationen erfolgen, die sich über Generationen akkumulieren. Einzelne Wissenschaftler auf dem Gebiet der evolutionären Entwicklungsbiologie versuchen also mitunter Modifikationen an den bestehenden Fundamenten der Evolutionsbiolgie zu erreichen, was auf heftige Kritik (auch aus den eigenen Reihen) stößt. Wahrscheinlich ist Evo-Devo daher eines der lebendigsten Forschungsgebiete in der aktuellen Biologie.
Die von mir im Folgenden besprochenen Studie untersuchen beide dieses zentrale Dogma von Evo-Devo genauer und lassen meines Erachtens auch Schlüsse zur genannten Kontroverse zu.
Cretekos et al. benutzen dazu die Unterschiede im Wachstum der Vorderextremitäten zwischen
Carollia perspicillata (einer Fledermaus) und der
Maus. Beide Taxa stammen aus unterschiedlichen Ordnungen der Säugetiere
Chiroptera (=Fledertiere) beziehungsweise Rodentia (=Nagetiere), teilen also einen gemeinsamen Vorfahren vor etwa 80-100 Millionen Jahren.
Die Studie
betrachtet Veränderungen in
Prx1, einem Transkriptionsfaktor, der durch klassische entwicklungsbiologische Methoden als wichtig in der Extremitätenentwicklung
identifiziert wurde. Die Kollegen stellten beim Verglich der Gen-Sequenzen aus Maus und Fledermaus nur einen nicht-synonymen (in das Protein übersetzten)Unterscheid fest. Dieser Unterschied befindet sich in einem Bereich des Genes, der ohnehin wenig konserviert ist, und nicht mit der typischen Funktion des Traskriptionsfaktors in Verbindung gebracht wird.
Unterschiede in der Expression von
Prx1 konnten in der späten Entwicklung der Vorderextremitäten festgestellt werden, in der Fledermaus war der Transkriptionsfaktor speziell im Bereich der Handwurzelknochen stärker exprimiert. Dieses Ergebnis korreliert gut mit dem zu diesem Zeitpunkt verstärkt auftretenden Längenwachstum in der Fledermaus.
Cretekos et al. betrachteten weiterhin also einen Enhancer "stromaufwärts" des Gens, der ebenfalls mit
klassischen Methoden identifiziert worden war. Dieser Enhancer enthält zwei Bereiche die zwischen Nager und Fledertier relativ konserviert sind, in diesen wurde dann die Funktion vermutet. Um dies zu testen konnte die Gruppe die jeweilige Enhancer-Region an ein
Reportegen koppeln und in Mäuse einbringen, dabei wurde stärkere Reporter-expression beim Chiroptera-Enhancer beobachtet.
Doch damit nicht genug, der Gruppe gelang es schließlich das Fledermaus-Kontrollelement in die Maus einzubringen, so dass es die Expression des
Prx1-Gens steuert. Die entsprechenden Mäuse zeigten tatsächlich ein verstärktes Wachstum der Vorderextremitäten.
Die Studie konnte so eindrucksvoll das zentrale Dogma der evolutionären Entwicklungsbiologie bestätigen und weiterhin zeigen, dass die zugrunde liegenden Veränderungen im untersuchten Fall auf Enhancer-Elementen basieren.
Auch die Studie von Prabhakar et al. beschäftigt sich mit der Funktion von Enhancern, die betreffende genomische Region war aber mit anderen Mitteln identifiziert worden. Dabei wurden komplett sequenzierten Genome von Wirbeltieren nach konservierten, nichtkodierenden Sequenzen durchsucht. Aus diesen Sequenze wurden wiederum jene
identifiziert, die in der Menschlichen Linie (entgegen des allgemeinen Trends) evolvieren. Diese Vorgehen basiert auf der Tatsache, dass Elemente mit einer bestimmten Funktion weniger evolvieren als funktionslose Elemente, ändern sie allerdings ihre Funktion erfolgt die Evolution sogar schneller als dies unter Neutralität (Funktionslosigkeit) der Fall wären. Die identifizierte Region liegt im
Intron eines Gens, das mit der Funktion des
Endosoms in Verbindung gebracht wird. Weier "stromäbwärts" befindet sich wieder ein Transkriptionsfaktor, dessen Wirkung die Entwicklung der Gliedmaßen
beeinflusst. Die Expression welcher Gene genau der mögliche Enhancer beeinflusst ist also noch nicht geklärt.
Zur Untersuchung der Funktion der Enhancer-Region benutzten die Kollegen also wieder ein Reporterassay (ß-Galactosidase). Sie brachten die postulierten Kontrollelemente aus
Rhesusaffen (Macaca mulatta),
Schimpansen (
Pan troglodytes) und dem
Menschen (
Homo sapiens) (Divergenz vor 6 bzw. 25 Mya) gekoppelt an das Reportergen, in Mäuse ein. So konnten die Forscher zeigen, dass die menschlichen Elemente im Vergleich zu denen aus beiden anderen Primaten, verstärkt Gene beim Wachstum der Extremitäten anschalten. Sie konnten so die durch den Genom-Verglich identifizierten Unterschiede bestätigen: Der Zustand und die Wirkung des Enhancers sind sich in nicht-menschlichen Affen ähnlich und entsprechen daher wahrscheinlich dem Zustand im gemeinsamen Vorfahren.
Mit sehr cleveren Experimenten bewiesen die Kollegen, dass genau die 13 Basen Unterschied im Menschen im Vergleich zu Schimpanse und Rhesusaffe den Unterschied ausmachen. Sie konstruierten Fragmente aus den beiden "Vierbeinern" in denen nur die betroffenen 13 Unterschiede eingebracht waren. Diese Konstrukte hatten die gleiche Wirkung wie das original-menschliche Element.
Die Studie konnte so, aufbauend auf dem zuvor beschriebenen zentralen Dogma von Evo-Devo, zeigen, dass durch Genomvergleiche die betreffenden Elemente identifiziert werden können. Ein denkbarer Name für ein solches Vorgehen ist "Vergleichende Entwiklungsgenomik" oder im Englischen "Comparative developmental genomics".
Weiter demonstrieren beide Studien eindrucksvoll die evolutionären Möglichkeiten für graduelle Veränderungen in Entwicklungsprogrammen. In beiden Beispielen konnte durch Veränderung einzelner Basen in regulativen Bereichen Unterschiede in der Genexpression erzeugt werden. Da es sich um Variationen in einzelnen Basenpaaren (
SNPs) handelt, die langsam und nacheinander ins Genom einfließen, verlangen die hier beobachteten Unterschiede geradezu nach Gradualismus.
Fee hat vor einigen Wochen auf dem Blog Science-meets-spciety ebenfall über das letztere Paper geschrieben und ich möchte einige Fehler in diesem (auch auf Researchblogging erschienenen)
Post abschließend korrigieren. Ich hoffe so eine
Diskussion anzuregen, falls es im deutschsprachigen Raum genügend Interesse gibt.
S-M-S:
Wir teilen bis zu 98% der codierenden DNA mit unseren nächsten Verwandten, den Schimpansen (Pan troglodytes) und doch unterscheiden wir uns markant von ihnen.
Das stimmt nicht! Eine der grundlegendsten Entdeckungen der
letzten Jahre waren Unterschiede in der
Kopienzahl einzelner Gene im Menschlichen Genom. 2007 wurde so (Hauptsächlich durch den Vergleich der Genome von Venter und Watson) offensichtlich, dass einzelne Menschen sich in 2-3% ihres Genoms unterscheiden. Der Unterschied zu unseren nächsten interspeziefschen Verwandten dürfe daher mindestens 5% betragen.
S-M-S:
Der größte Teil der DNA in menschlichen Zellen ist nicht kodierend und wurde, als man dies entdeckte, fälschlicherweise als Junk-DNA (Abfall-DNA) verschrien.
Diese Sichtweise ist grundsätzlich falsch! Es wurden
seit der Entdeckung nicht-kodierender Bereiche schon versucht Funktionen für diese für eine höhere Organisation zu postulieren. Vergleicht man aber unterschiedliche Organismen (z.B. der
Zwiebel oder des Salamanders) mit der des Menschen oder der
Kugelfische (
Tetraodontidae; anderes Extrem) fällt schnell auf dass eine höhere Komplexität
nicht mit der
Genomgröße korreliert.
Die in Frage stehenden regulatorischen Elemente machen einen winzigen Teil des Genoms aus.
S-M-S:
Dabei stiessen sie auf einen Abschnitt von 546 Basenpaaren Länge, der sich seit der Entwicklung der Wirbeltiere nur wenig verändert hatte. Jedoch hatten sich in der relativ kurzen Zeit von 6 Millionen Jahren, seit sich die Entwicklungszweige von Mensch und Schimanse trennten, 16 Veränderungen etabliert, die alle in einem Abschnitt von 81 Basenpaaren clusterten. So etwas ist für einen genetischen Detektiv ein eindeutiges Indiz, dass weitere Untersuchungen gewinnbringend sein könnten.
Die Forscher wussten im Gegensatz zum Schreiber dieser Zeilen um die Existenz von Enhancen. Andernfalls hätten sie den entsprechenden Bereich nicht als solchen identifizieren können. In der besprochenen Studie wurden nicht zum ersten Mal Selektion auf einen nicht-kodierenden Bereich nachgewiesen. Ähnliche Fehlinterpretationen und die übertriebene Darstellung von Neuheiten (wissenschaftlichen Revolutionen) in der Wissenschaftsberichterstattung veranlassen auch beispielsweise Kreationisten regelmäßig zu
ähnlichen Dummheiten.
S-M-S:
[...]so aktivierten alle die Expression von Genen in den Augen, Ohren und in den embryonalen Kiemenbögen, die später den Kiefer bilden.
Die Expression eines Reportergens! Dies ist im Vergleich zu den anderen Fehlern aber eher zweitrangig.
S-M-S:
Ein neuer Teilbereich der entwicklungsgenetischen Forschung, der bestimmt noch viele Überraschungen und neue Erkenntnisse birg.
Quatsch! "Vergleichende Entwicklungsgenomik" ist zwar ein recht neuer boomender Bereich der Entwicklunsbiologie, erfunden wurde er aber in diesem Paper nicht. Die Studie bestätigt vielmehr experimentell die Validität der zugrundeliegenden
in silico Analysen.
Ich hab hier mal die jährliche Zähl an Veröffentlichungen, die "Comparative developmental genomics" im Volltext (Pub-med) erwähnen geplotet:
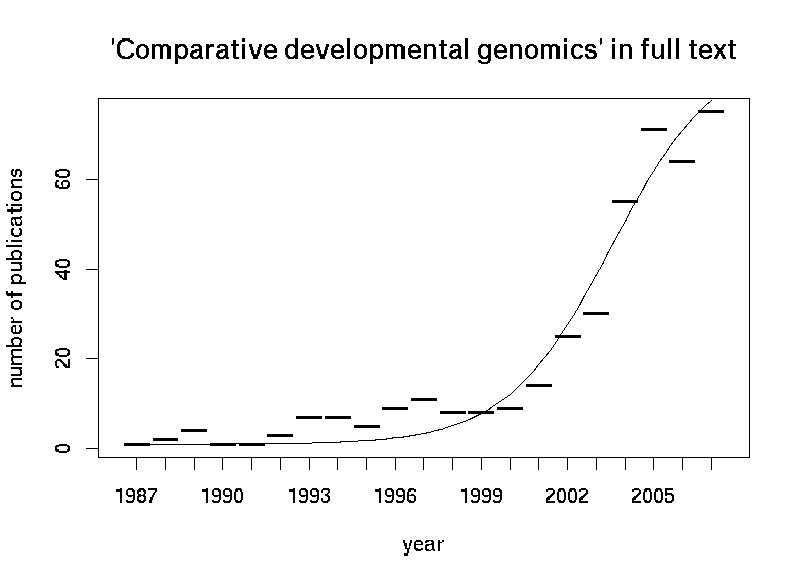
Dabei fällt auf, dass
das erste Paper bereits aus dem Jahre 1988 stammt, als eigentlich noch keine Genome zum Vergleich bereitstanden. Veröffentlichungen vor dem Jahre 2001 können also wahrscheinlich als "Hintergrundrauschen" oder "Vorahnung" interpretiert werden, wirkliche "Comparative genomics" in dem heute etablierten Sinn waren damals noch kaum möglich. Ab diesem Zeitpunkt wurden dann anhand der vorhandenen Daten die betreffenden Methoden entwickelt. Prabhakar et al. haben also zwar eine schöne Studie angefertigt, mitnichten aber das "Teilgebiet" erfunden.
Kommentare/ Kritik erwünscht, ich bin kein Entwicklungsbiologe und habe sicher selbst Fehler gemacht!
________________________________________________________________________________________________________________
C. J. Cretekos, Y. Wang, E. D. Green, J. F. Martin, J. J. Rasweiler, R. R. Behringer (2008). Regulatory divergence modifies limb length between mammals Genes & Development, 22 (2), 141-151 DOI: 10.1101/gad.1620408S. Prabhakar, A. Visel, J. A. Akiyama, M. Shoukry, K. D. Lewis, A. Holt, I. Plajzer-Frick, H. Morrison, D. R. FitzPatrick, V. Afzal, L. A. Pennacchio, E. M. Rubin, J. P. Noonan (2008). Human-Specific Gain of Function in a Developmental Enhancer Science, 321 (5894), 1346-1350 DOI: 10.1126/science.1159974



